(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Mitochondrial disorders encompass a highly heterogeneous group of diseases characterized by not only defects in the OXPHOS system but also alterations in diverse signaling pathways, intracellular Ca2+ homeostasis, and cell survival and death. Numerous pathogenic mutations in genes encoding proteins, in genes involved in mtDNA synthesis and maintenance, OXPHOS subunits and complex assembly factors, and proteins implicated in mitochondrial dynamics have been described. Growing number of identified disease-causing mutations in both nuclear and mitochondrial genomes makes diagnosis of mitochondrial disorders extremely challenging. The current algorithm for molecular diagnosis of patients with suspected mitochondrial disease is a complex multistep process that implicates thorough clinical testing, histochemical and biochemical analyses, followed by molecular genetic screening.
The advent of the “omics” era has opened up unprecedented opportunities for a system biology approach to assess mitochondrial function and to diagnose mitochondria-related cardiovascular disease (CVD). The development of next-generation sequencing (NGS) and global gene expression profiling technologies has enabled genome-wide screening for disease-related mutations, single nucleotide polymorphisms, and copy number variants in time- and cost-effective manner. While progress in proteomics has not been as dramatic, recent technological advancements in this field have enabled the identification of proteins at sub-femtomolar levels in complex biological samples and brought quantification to the proteome accelerating translation of proteomic research into clinical practice.
Despite the enormous potential of this systemic approach, its use for diagnosis of mitochondria-related cardiac diseases remains rather limited. The convergence of mitochondrial research and high-throughput genomic, transcriptomic, proteomic, and bioinformatic technologies will improve our understanding of the role of this organelle in the heart and will ultimately lead to more individualized targeted therapies of mitochondria-related CVD.
Introduction
Over the past decades, due to great advances in molecular and cell biology, biochemistry, and genetics, our view on mitochondria as a relatively static cellular powerhouse has radically changed. It is now well appreciated the central role of the organelle as a highly dynamic integrator of energy production and diverse signaling pathways, intracellular Ca2+ homeostasis, and cell survival and death. Although all these functions are essential for virtually all human organs, they are of particular importance for the heart to maintain the permanent rhythmic contractions and ensure oxygenation of all tissues of the body.
Mitochondrion is the only intracellular organelle that possesses its own genome, mtDNA. Human mtDNA encodes 13 subunits of the ETC complexes, 2 rRNAs, and 22 tRNAs, while the majority of ∼1,500 mitochondrial proteins are encoded by nuclear genomes [1, 2]. However, clinical researchers for some time have ignored the role of defects in mitochondrial genes in the pathogenesis of human diseases. The clinical study by Luft et al. [3] showing abnormalities of mitochondrial morphology and function in muscle of a patient with severe hypermetabolism of nonthyroid origin has introduced the concept of mitochondrial disorders and mitochondrial medicine [4, 5]. However, only more than two decades later, in 1988, it was found that mutations in mtDNA were associated with human diseases, myopathies, and optic neuropathy, and the molecular era of mitochondrial medicine has begun [6, 7].
While mitochondrial diseases have initially been considered to be rare disorders, recent studies estimate that approximately 1 in 5,000 children will develop a mitochondrial disorder [8, 9]. Furthermore, approximately 1 in 200 adult individuals carry mtDNA mutations and can transmit mitochondrial diseases [10, 11].
Mitochondrial disorders are genetically heterogeneous as they can be caused by mutations in both mitochondrial and nuclear genomes. Mitochondrial diseases display also very variable clinical features; neuromuscular abnormalities are generally the main symptoms, including seizures, skeletal muscle weakness, cardiomyopathy, exercise intolerance, optic atrophy, hearing loss, diabetes mellitus, renal and gastrointestinal defects, as well as immunodeficiency [12, 13].
Due to the highly diverse and multisystem nature of mitochondrial disorders, their diagnosis represents an extremely challenging task [14, 15]. To address this critical issue, the whole arsenal of modern molecular and cell biology and genetic technologies has intensively been exploited. They range from PCR, restriction fragment length polymorphism (RFLP), and single-strand conformation polymorphism (SSCP) analyses to high-throughput analytical techniques, such as capillary array electrophoresis and denaturing high-performance liquid chromatography. In addition, super-resolution fluorescence microscopy and high-resolution scanning electron microscopy and tomography as well as noninvasive magnetic resonance spectroscopy, large-scale mitochondrial proteomic, and protein array analysis are being recruited to uncover the molecular basis of the heterogeneous mitochondrial disorders associated with cardiovascular disease (CVD).
Recently, emerging mitochondrial gene profiling by “high-density” (>10,000 genes) microarrays (cDNA and oligonucleotides) may allow for simultaneous assessment of the expression of thousands of genes encoding mitochondrial proteins [16–19]. Combined with bioinformatic analysis, this approach provides a revolutionizing opportunity to identify novel markers for diagnosis of mitochondrial disorders and facilitates the development of efficient therapy.
mtDNA Mutations
mtDNA is characterized by significantly higher (10- to 20-fold) mutation rate compared to its nuclear counterpart [20–23]. It is believed that major factors that contribute to higher mtDNA vulnerability are the lack of protective histones, its close proximity to OXPHOS complexes producing ROS, and inefficient mtDNA repair.
Since the first identification of mutations in mtDNA associated with human diseases in 1988, to date more than 300 pathogenic mtDNA mutations have been reported (http://www.mitomap.org). Mutations occur throughout the mitochondrial genome; however, a region of increased mutability within the D loop has been identified, whereas mutations in rRNA genes are less frequent. Both point mutations and rearrangements, mainly large-scale (1.3–8 kb) deletions spanning several genes, have been found (Fig. 24.1).
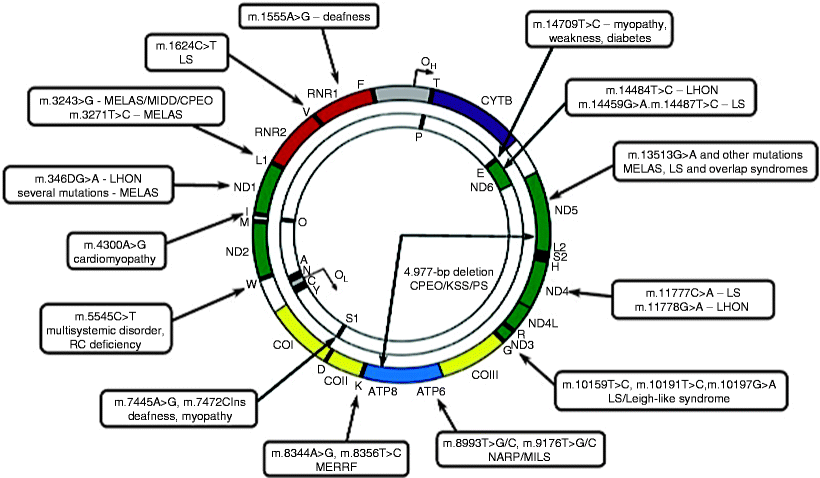
Fig. 24.1
Schematic representation of mtDNA mutations causing mitochondrial diseases. A map of the human mitochondrial genome with sites of common mtDNA mutations and associated disorders is shown. CPEO chronic progressive external ophtalmoplegia; LHON Leber hereditary optic neuropathy; LS Leigh syndrome; MELAS mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes; MERRF myoclonic epilepsy and ragged red fibers; MILS maternally inherited Leigh syndrome; NARP, neurogenic weakness, ataxia, and retinitis pigmentosa; PS Pearson syndrome (adapted from Tuppen et al. [24] with permission from Elsevier)
While mtDNA point mutations occur within the genes encoding proteins, rRNAs, and tRNAs, >50% of pathogenic mutations have been identified in mt-tRNAs. One of the most common mtDNA point mutations is the m.3243A >G mutation in tRNALeu(UUR), which causes mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome or diabetes, hearing loss, and retinopathy [25]. Another common point mutation, m.8344A >G in tRNALys, causes myoclonic epilepsy with ragged red fibers (MERRF) syndrome [26]. The genes encoding tRNALeu(UUR) and tRNALys represent mutation hot spots within which several disease-causing mutations have been identified (Fig. 24.1).
The m.1494 T >C and m.1555A >G mutations are point mutations in the mitochondrial 12S rRNA gene associated with deafness. Examples of disease-causing mutations in the genes encoding proteins include m.8993 T >C and m.8993 T >G in the ATP6 gene, which cause maternally inherited Leigh syndrome (MILS) and neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome, respectively, and m.3460 G >A, m.11778 G >A, and m.14484 T >C in the ND1, ND4, and ND6 genes, respectively, which cause Leber hereditary optic neuropathy (LHON) (Fig. 24.1) [7, 27, 28].
Finally, large-scale deletions in mtDNA have been identified to be associated with myopathy, chronic progressive external ophthalmoplegia (CPEO), Kearns-Sayre syndrome (KSS), and Pearson syndrome (PS) (Fig. 24.1) [6, 29, 30]. Currently, more than 120 deletions in mtDNA have been reported in association with various pathologies (http://www.mitomap.org). Although the precise mechanism of their generation is not known, they occur more frequently in mtDNA regions flanked by tandem repeats, and recombination repair of mtDNA has been suggested to be implicated in deletion formation [31, 32].
nDNA Mutations
Given the vast majority of approximately 1,500 mitochondrial proteins including most of OXPHOS subunits encoded by nuclear genome, it is not surprising that the list of nDNA mutations associated with different mitochondrial disorders is constantly growing. These mutations can be subdivided into several groups according to the processes, which mediate affected mitochondrial proteins (Table 24.1) [5, 15, 33].
Table 24.1
Mutated nuclear genes associated with mitochondrial diseases
Affected process | Mutated gene |
|---|---|
mtDNA synthesis and maintenance; nucleotide metabolism | POLG1, POLG2, TWINKLE, DGUOK, MPV17, RRM2B, SUCLA2, SUCLG1, SUCLG2, TK2, TYMP |
mtDNA transcription | GABPA, GABPB1, NRF1, PGC1, TFAM |
mtDNA translation | DARS2, MRPS16, MRPS22, MRSL33, PUS1, RARS2, TSFM, TUFM |
OXPHOS subunits | COX6B1, COX4I2, CYCS, NDUFA1, NDUFA2, NDUFA11, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS5, NDUFS6, NDUFS7, NDUFS8, SDHA, SDHB, SDHC, SDHD |
OXPHOS assembly factor | ATP12, BCS1L, COX6B2, COX10, COX15, LRPPRC, NDUFAF1, NDUFAF2, SCO1, SCO2, SDH5, SURF1, TMEM70 |
Mitochondrial dynamics | DRP1, GDAP1, KIF5A, LETM1, MFN2, OPA1, OPA3, PARK2, PINK1 |
Mitochondrial transport | ANT1, CACT, DNAJC19, HSPD1, PHC, TIMM8A, VDAC |
Coenzyme Q10 biosynthesis | CABC1, COQ2, COQ9, PDSS1, PDSS2 |
Cardiolipin biosynthesis | TAZ |
Others | ABCB7, APTX, ASS1, BCKDHA, BCKDHB, C12orf65, C14orf156, C18orf22, CPS1, CPT1A, CPT1B, CPT2, DLD, ETFA, ETFB, ETFDH, FASTKD2, FH, HADHA, HADHB, HLCS, HMGCL, MCCC1, MCCC2, MMAA, MMAB, MMACHC, MMADHC, MRP63, MUT, NAGS, OGDH, OTC, OXA1L, PC, PCK2, PDHA1, PDHB, PDHX, PDK1, PPM1B |
Defects in mtDNA Dynamics
Mutations in the POLG1 gene encoding mitochondrial DNA polymerase γ represent one of the most common mitochondrial diseases responsible for Alpers’ syndrome [34–36]. This disorder can be inherited in dominant or recessive form. The clinical phenotypes of POLG1 deficiency are very heterogeneous and include severe forms of childhood myocerebrohepatopathies, sensory ataxic neuropathy, spinocerebellar ataxia with epilepsy, and progressive external ophthalmoplegia (PEO) [37, 38]. Mutations in the accessory subunit gene of DNA polymerase γ, POLG2, have been found in a dominant form of PEO [39].
More than 30 different mutations in the TWINKLE (also known as PEO1) gene, which encodes the replicative DNA helicase Twinkle, have currently been identified [40]. The TWINKLE mutations result in depletion of mtDNA leading to mitochondrial dysfunction and are associated with neuromuscular abnormalities. All dominant TWINKLE mutations cause autosomal dominant PEO characterized by exercise intolerance and muscle weakness; the ocular muscles are most severely affected [41, 42]. Autosomal recessive TWINKLE mutations are responsible for a hepatocerebral form of mtDNA depletion disorder [43].
In addition to TWINKLE and POLG1 mutations, multiple mutations in the DGUOK, MPV17, RRM2B, SUCLA2, SUCLG, TK2, and TYMP (also known as ECGF1) genes also lead to the mtDNA depletion syndromes [15, 44–49]. MPV17 encodes a mitochondrial inner membrane protein with unknown yet function, while the other genes encode proteins implicated in the maintenance of the mitochondrial dNTPs pool. Mutations in another gene belonging to this group, ANT1, encoding the adenine nucleotide translocator 1, have been identified to be associated with autosomal dominant PEO [50].
Defects in mtDNA Translation
Defects in any component of mtDNA translation apparatus, including mitochondrial translation factors, ribosomal proteins, amino acyl-tRNA synthetases, and tRNA posttranslation modification enzymes, could lead to mitochondrial disorders [15]. These types of mitochondrial diseases are typically characterized by OXPHOS deficiencies, lactic acidosis, and myopathy [51, 52]. Currently, mutations in genes, which encode mitochondrial ribosomal proteins MRPS16 and MRPS22, pseudouridine synthase 1, mitochondrial translation elongation factors, and arginyl- and aspartyl-tRNA synthetases, have been found [53–62].
Defects in OXPHOS Complexes
In 1995, the first mutation in a nuclear succinate dehydrogenase gene encoding the flavoprotein subunit of Complex II was detected in two sisters with Leigh syndrome [63]. Then, multiple mutations in subunits of Complex I in patients with Leigh or Leigh-like syndromes have been identified [64–68]. A deletion in the gene QP-C encoding a subunit of Complex III has been found in a patient with hepatomegaly and hypoglycemia [69]. Mutations in the nuclear genes, which encode subunits of Complex IV (cytochrome c oxidase, COX), appear to be very rare; however, the first mutation in COX6B1 has recently been shown to be associated with severe infantile encephalomyopathy [70]. So far no mutations in nuclear genes encoding the 12 subunits of ATP synthase (Complex V) have been found.
In addition to mutations in OXHOS subunits, multiple mutations in nuclear genes encoding complex assembly factors have been shown to be responsible for various mitochondrial diseases. They include mutations in Complex IV assembly factor genes: SCO1 and SCO2 associated with encephalohepatopathy and cardiomyopathy, respectively, in SURF1 associated with Leigh syndrome, in COX10 and COX15 found in patients with encephaloneuropathy and myopathy, respectively [71–74]. Furthermore, mutations in the BCS1L gene, encoding an assembly factor of Complex III, and in ATP12 and TMEM70, encoding Complex V assembly factors, have been identified [75–78].
Defects in Mitochondrial Dynamics
Emerging evidence of the critical role of mitochondrial dynamics in human health and disease has been discussed in Chapter 17. Numerous mutations in the genes encoding proteins involved in mitochondrial fusion and fission, such as MFN2, OPA1, DRP1, GDAP1, and LETM1, have been reported to be associated with inherited neurodegenerative disorders. They include Charcot-Marie-Tooth neuropathy, autosomal dominant optic atrophy, abnormal brain development, and the Wolf-Hirschhorn syndrome. Moreover, several loss-of-function mutations in PINK1 and PARK2, encoding PINK1 and Parkin, respectively, which mediate mitochondrial trafficking, cause early-onset Parkinson’s disease.
Defects in Other Nuclear Genes Controlling Mitochondrial Functions
Molecular defects in acetyltransferase taffazin, implicated in metabolism of cardiolipin, an abundant phospholipid in the mitochondrial inner membrane, have been demonstrated [79, 80]. Given a key role of cardiolipin in mitochondrial function, it is not surprising that mutations in the TAZ gene, which encodes taffazin, cause severe OXPHOS defects and contribute to the pathogenesis of Barth syndrome.
Most mitochondrial proteins are encoded by nDNA, synthesized in the cytosol, and must be imported to mitochondria by the action of the complex mitochondrial import machineries [81]. Several mutations in the genes encoding proteins involved in mitochondrial protein import have been identified. Affected genes include TIMM8A, defect is associated with the X-linked Mohr-Tranebjaerg syndrome; HSPD1, defect is associated with brain hypomyelination and leukodystrophy; and DNAJC19, defect is associated with an autosomal recessive dilated cardiomyopathy with ataxia [82–85].
Models for Mitochondrial Disorders
Despite great advances in our understanding of mitochondrial structure and function and their key role in heart physiology and pathophysiology, the development of suitable experimental models for human mitochondrial diseases represents significant technical challenge. To date, direct manipulation of mtDNA remains problematic due to the inability to transfect plasmids or engineered mtDNAs into mammalian mitochondria [91]. Another difficulty is related to the polyploidy of the mitochondrial genome: more than 1,000 of mtDNA molecules are present within a cardiomyocyte.
Yeast Models
Baker’s yeast, Saccharomyces cerevisiae, is an ideal model eukaryotic organism to study the molecular mechanisms underlying mitochondrial function, biogenesis, and dynamics due to the great genetic and biochemical tractability of the obtained data. At present, S. cerevisiae represents the only experimental model organism, in which transformation of mtDNA can be achieved and mutations can be introduced in specific mtDNA genes via homologous recombination [92]. Importantly, S. cerevisiae survives in the absence of mtDNA providing thereby a unique possibility to examine the most severe mitochondrial defects. Hence, yeast experimental models have been extensively used to study human mitochondrial pathogenic mutations, including MELAS mutations, mt-tRNA and leucyl-tRNA synthetase mutations, and MT-ATP6 mutations [93–98]. However, there are some limitations in using of S. cerevisiae as a model for human mitochondrial disorders [24].
Cytoplasmic Hybrid (Cybrid) Models
Introduction of human transmitochondrial cytoplasmic hybrid cells, cybrids, based on fusion of enucleated cells derived from patients with human cell lines lacking endogenous mtDNA, so-called ρ0 cells, has been a major breakthrough in the development in vitro models for studying mitochondrial diseases [99]. This experimental tool provides the opportunity to determine the genetic cause of a mitochondrial disease, to study the molecular mechanisms, by which the genetic defect affects cellular metabolism, and analyze the consequences of variations in the levels heteroplasmy of mtDNA mutations (Fig. 24.2) [100, 101].
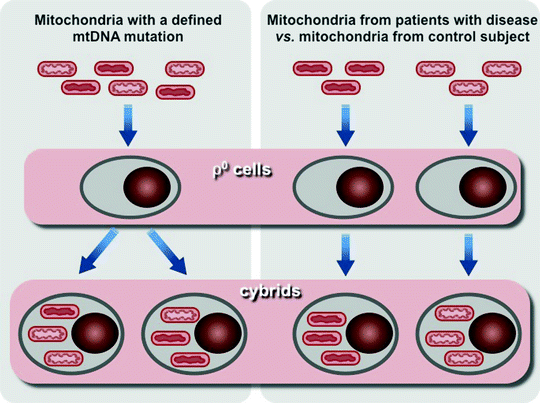
Fig. 24.2
Scheme of two major approaches to use human transmitochondrial cytoplasmic hybrid cells (cybrids). Left panel, mitochondria containing mtDNA with a known heteroplasmic mutation is transferred into cells depleted of their own mtDNA (ρ0 cells) to generate cybrids. Resultant cybrids after expansion can be analyzed to assess the effects of the mutation on cellular phenotype and the consequences of variations in the levels heteroplasmy of mtDNA mutation. Right panel, mitochondria containing uncharacterized mtDNA mutation from a patient and mitochondria from a control subject are used to generate two sets of cybrids. Comparison of these two sets of cybrides allows to analyze differences caused by mutant mtDNA
However, the cybrid technology has a few drawbacks that have to be taken into account. Aneuploid tumor cells are typically used for the generation of cybrids. Unlike the majority of human cells, which energy metabolism relies mainly on OXPHOS, bioenergetics of tumor cells depends on anaerobic glycolysis. Moreover, the aneuploid nature of tumor cells can change the ratio of mtDNA- and nDNA-encoded subunits of OXPHOS complexes and thereby may affect mitochondrial function. In addition, microarray analyses have shown that preparation of ρ0 cells and cybrids result in significant changes in expression of multiple genes, including nuclear OXPHOS genes [102, 103]. These features of cybrid systems might be responsible for observed differences between in vivo and in vitro cybrid phenotypes. It has recently been demonstrated that the pathogenic mutation in the MT-IV gene (m.1624 C > T) associated with significantly decreased activities of Complex I and IV and elevated neonatal lethality, but when introduced in cybrids, it has no effect on mitochondrial respiration [104].
Mouse Models
Although yeast and cybrid model systems are important experimental tools for studying mitochondrial disorders, the development of mouse models are vital for progress in this field.
Transmitochondrial Mice
Since direct engineering of mtDNA remains challenging, more indirect approaches have been used for generating mouse models harboring mtDNA mutations. These approaches are based on cell biology techniques to transfer modified mtDNAs into fertilized mouse eggs. Enucleated cells (cytoplasts) can be fused with zygotes or cytoplasts can be fused first with embryonic stem cells followed by their injection into blastocysts.
In 1996, the first heteroplasmic mouse model was created using cytoplasmic fusion of two normal inbred mouse strains with distinct haplotypes [105, 106]. Generated heteroplasmic mice carry only polymorphic mtDNAs and display different levels of heteroplasmy. Interestingly, tissue-specific selection for a certain mtDNA genotype in some tissue has been observed; this directional selection has been dependent on tissue-specific single factors [107, 108]. Although these mice have not shown significant pathological changes, these models can be used in search for proteins mediating tissue specificity and for deciphering the molecular mechanisms underlying mtDNA somatic segregation.
Cytoplast fusion with zygotes has been used to generate the “mito-mice” model carrying a heteroplasmic mtDNA deletion. The “mito-mice” have displayed Complex IV deficiency associated with myopathy and cardiomyopathy, renal failure, anemia, deafness, lactic acidosis, and a shortened life-span [109, 110]. The mutant phenotype resembles the clinical presentation of early-onset Pearson syndrome caused by a single heteroplasmic mtDNA deletion.
Recently, a similar approach has been used to generate transmitochondrial mice harboring heteroplasmic point mutations in the MT-COX1 (m.6589 T > C) and MT-ND6 (m.13885insC) genes. Importantly, these mice have developed mitochondrial myopathy and cardiomyopathy [111]. Moreover, transmitochondrial mice created by the injection of mutant (m.3234A > C) mitochondria into the C57Bl/6 mouse zygotes have been characterized by severe mitochondrial dysfunction associated with elevated ROS generation in heteroplasmic tissues [112].
Cytoplast fusion with ESCs, which have been transmitted to the C57Bl/6 mice, has been exploited to generate transmitochondrial mouse models carrying mutations in the 16S rRNA or MT-COX1 (m.6589 T > C) genes [113–115]. Mutant mice have OXPHOS deficiency, myopathy and cardiomyopathy, lactic acidosis, and growth retardation.
Mouse Models with Targeted Nuclear Mitochondrial Genes
In contrast to significant technical difficulties in generating mouse models with mtDNA mutations, the manipulation of nuclear genes encoding mitochondrial proteins has been more successful. Constantly growing list of such mouse models is summarized by Vempati U.D. et al. [116]. Knockouts of structural subunits and assembly factors of OXPHOS complexes I–IV have successfully been created. They include Ndufs4 and AIF (Complex I), SDHD (Complex II), RISP (Complex III), COX VIaH, COX10, Cyt c s , Cyt c t , and Surf1 (Complex IV) knockout mice. Multiple transgenic mice have been generated with targeted disruption of genes that control the maintenance of mtDNA and nucleotide pool, such as PolgA (“Mutator” model), Tfam, Twinkle (“deletor” model), Mterf3, Ant1, Rrm2b, and Tk2 [116].
It is important to note that several mouse models for mitochondrial disorders show signs of cardiac diseases. Transgenic mice with the AIF deletion in the heart and skeletal muscle have displayed mitochondrial dysfunction and developed skeletal muscle atrophy and severe dilated cardiomyopathy leading to heart failure (HF) [117]. COX VIaH −/− mice have exhibited significant decrease in Complex IV activity associated with impaired ventricular filling and diastolic dysfunction [118].
Transgenic mice with the cardiac-specific Tfam deletion have exhibited decreased levels of mtDNA and mtDNA encoded proteins as well as reduced activities of Complex I and IV in the heart. Moreover, Tfam −/− mice have dilated cardiomyopathy with atrioventricular conduction blocks resulting in 75% embryonic lethality [119, 120]. Two transgenic mice carrying pathogenic Twinkle mutations, a point mutation 360A >T and a duplication of amino acid residues 353–365 found in patients, have been generated. The mutant animals have had multiple deletions in mtDNA in the heart and in brain, but not in the skeletal muscle, while a modest decrease in COX activity has been observed in both brain and muscle [121]. Cardiac-specific deletion of the Mterf3 gene, encoding one of the mitochondrial transcription termination factors, has caused an increase in mtDNA transcription initiation and impaired mitochondrial function associated with mitochondrial cardiomyopathy and shortened life-span [122].
Loss of mitochondrial inner membrane protein Ant1, which mediates ATP transport from mitochondria to the cytosol, has resulted in mitochondrial myopathy and cardiomyopathy [123]. Interestingly, Ant1 −/− mice have also exhibited upregulation of some OXPHOS subunits, mt-rRNA, and mt-tRNA genes resembling changes found in patients with mitochondrial disorders [124].
Knockout of the mitochondrial free-radical scavenger Mn2+-superoxide dismutase (SOD2) has led to a drastic decrease in Complex II and TCA enzyme aconitase activities mostly in the myocardium. SOD2 −/− mice develop dilated cardiomyopathy, accumulate lipids in liver and skeletal muscle, and die within 10 days after birth [125]. Another SOD2 −/− mice have displayed oxidative damage of neuronal and cardiac mitochondria associated with severe anemia, neuronal degeneration, progressive motor impairments, or severe dilated cardiomyopathy depending on genetic background [126, 127]. The mutant mice have died within 18 days of life. Surprisingly, liver-specific SOD2 −/− mice have not displayed increased lipid peroxidation or other biochemical defects [128]. Similarly, motor neuron-specific SOD2 deletion has not resulted in significant increase in oxidative damage of the motor neurons, although their axons have been more sensitive to nerve injury [129].
Growing evidence suggests that tightly balanced mitochondrial fusion and fission (division) play a critical role in the high-energy demanding myocardium. Impairment of this balance represents a novel class of mitochondrial disorders contributed to the pathogenesis of various (CVD) (see Chap. 17). Recently, several transgenic mouse models with targeted genes, which control mitochondrial dynamics, have been generated. Cardiac-specific deletion of one of the two known mitofusins, Mfn1 or Mfn2, implicated in mitochondrial fusion, has caused a very modest cardiac hypertrophy [130]. However, conditional cardiac-specific deletion of both mitofusins, Mfn1 and Mfn2, has caused mitochondrial fragmentation, respiratory dysfunction in the heart leading eventually to lethal dilated cardiomyopathy [131]. Moreover, since Mfn2 plays a role in mitochondria-dependent apoptosis, mouse Mfn2-defficient cardiomyocytes have been resistant to apoptosis-inducing stimuli, and Mfn2 −/− mice have exhibited better recovery after postischemic reperfusion injury [130].
More recently, the first direct evidence that a mutation in the mouse mitochondrial fission gene Drp1 can mimic mitochondrial cardiomyopathy has been reported [132]. The Python point mutation (C452F) in a highly conserved region of the middle domain of Drp1 impairs its oligomerization, which is essential for Drp1 fission function [132–134]. Mice with homozygous Python mutation have died prenatally, whereas heterozygous Python myocardium has exhibited reduced mitochondrial ATP synthesis. Accordingly, fibroblasts harboring heterozygous Python mutation have displayed abnormal mitochondria and peroxisomes [132].
Diagnosis of Mitochondrial Disorders
Identification of responsible genes, specific disease-causing mutations, and the type of their inheritance is critically important for understanding the molecular mechanisms of mitochondrial diseases and choosing more specific and appropriate medical management of the patient. In addition, a precise molecular diagnosis enables genetic testing of members of at risk families, greatly improving genetic counseling and risk estimation for the family. Mitochondrial function depends on a complex interplay between mitochondrial and nuclear genomes, and its impairment can be caused by more than 300 pathogenic mutations in both mtDNA and nDNA. The lack of clear genotype-phenotype correlations in most mitochondrial disorders and the heteroplasmic nature of mtDNA further highly complicate the diagnosis of mitochondrial disease. Due to these characteristics of mitochondrial disorders, their precise diagnosis requires a multidisciplinary approach.
Current diagnostic algorithm typically involves clinical testing and evaluation of family history followed by thorough histochemical and biochemical analysis of clinically relevant tissues, for example, skeletal muscle or blood [33, 135, 136]. If such a screening shows mitochondrial dysfunction, then the application of molecular genetic techniques is warranted (Fig. 24.3) [135].
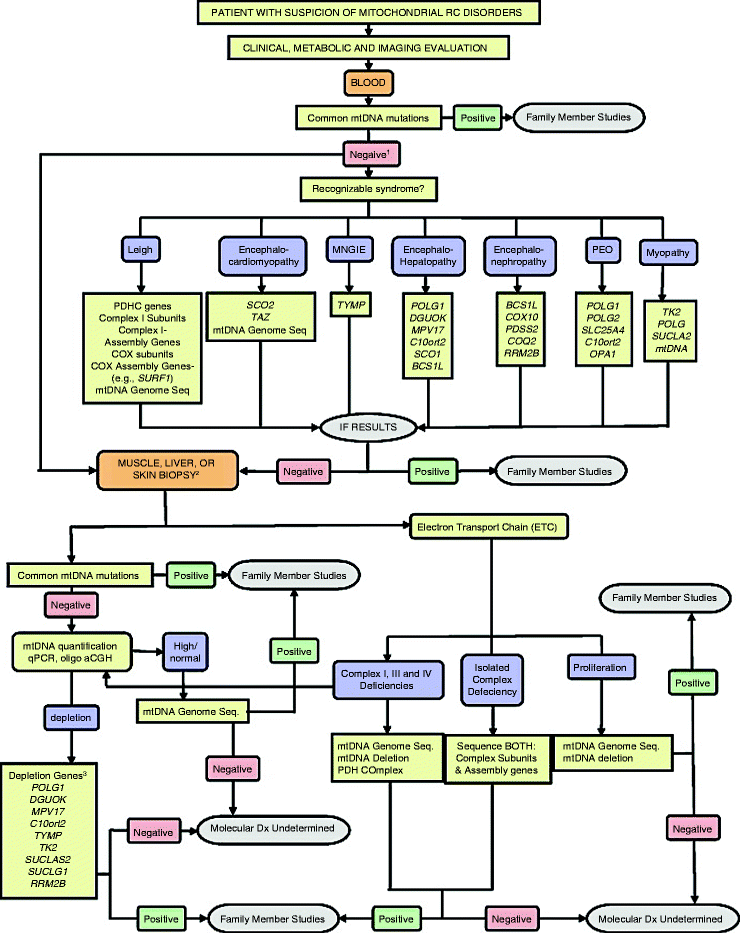
Fig. 24.3
Testing algorithm for molecular diagnosis of patients with suspected mitochondrial disease. A generalized algorithm for molecular testing based on clinical and biochemical information is presented. This algorithm is designed to be a general guide and is not intended to encompass every potential clinical scenario nor all possible genetic etiologies. For the genes listed, HUGO gene nomenclature is used. MNGIE, mitochondrial neurogastrointestinal encephalopathy syndrome; PEO, progressive external ophthalmoplegia syndrome; Oligo aCGH, oligonucleotide array-comparative genomic hybridization. (1) If a common mtDNA mutation is not detected in the initial screen, then a tissue biopsy for additional molecular and biochemical studies is required. If a tissue biopsy is not available, then screening appropriate nuclear genes based on a recognized clinical pattern can be pursued. (2) A muscle or liver biopsy can be used for both molecular (tissue-specific mtDNA mutations/heteroplasmy and/or mtDNA quantification) and biochemical studies (ETC activity measurements). A skin biopsy for fibroblasts may be appropriate for biochemical studies, especially in infants too young for muscle biopsy, but is not useful for detecting mtDNA depletion or multiple deletions of mtDNA. (3) POLG, DGUOK, MPV17, and C10orf2 mutations have been observed with mtDNA depletion and hepatoencephalopathy. TYMP mutations are associated with MNGIE. TK2, SUCLA2, SUCLG1, and RRM2B have been observed with mtDNA depletion and encephalomyopathy. Myopathy with elevated creatine kinase is a frequent feature of TK2 deficiency (from Wong et al. [135] with permission from Elsevier)
Histological and Biochemical Screening
Recent advances in imaging and biochemical techniques to identify changes in mitochondrial morphology and function associated with mitochondrial diseases have previously been discussed (see Chap. 2). Briefly, they include the use of various fluorescent dye markers and fluorescent imaging, and “super-resolution” fluorescence technologies allowing analysis of the fine details of mitochondrial structure in living cells [137–140]. High-resolution scanning electron microscopy, electron tomography, and cryo-electron tomography, combined with 3D imaging, visualize not only mitochondrial ultrastructure but also mitochondrial complexes at near-atomic resolution [141–144].
Two major in vitro approaches can be used to accurately assess the integral mitochondrial functional activity: a bioluminescent measurement of ATP production and polarographic measurement of oxygen consumption [145–148]. The development of high-resolution respirometry allows serial measurements of oxygen consumption in the same very small biopsy sample [149–151]. Moreover, using various substrates activities of different OXPHOS complexes can be estimated.
Noninvasive techniques based on magnetic resonance spectroscopy (MRS) have become popular in assessing mitochondrial function in vivo in various human tissues including the heart [152–156]. 13C-MRS measures the tricarboxylic acid (TCA) cycle, glycolysis, or β-oxidation, whereas31P-MRS assesses ATP synthesis and phosphocreatine dynamics. However, to date application of MRS in clinical practice is rather limited due to its insufficient reproducibility, low spatial and temporal resolution, and long acquisition times.
Highly sensitive spectrophotometric-based enzyme assays using small amounts of tissues allow to measure individual enzyme activities and evaluate the functionality of specific steps of mitochondrial respiration [147, 154, 157]. Combination of enzyme essays with immunohistochemistry using specific antibodies against individual OXPHOS subunits is particularly helpful. Such approach allows better detect mosaic mitochondrial defects than biochemical methods alone, which are typically performed with tissue homogenates [158, 159]. Detection of the defective OXPHOS complex or specific subunit will provide information for molecular genetic screening of mutations within corresponding genes. Reduced activities of OXPHOS complex I+III and complex II+IV suggest defects in genes controlling CoQ10 biosynthetic pathways (ADCK3, CABC1, CoQ2, CoQ9, PDSS1, or PDSS2), whereas deficiency in Complexes I, III, IV, and V may be caused by deletions in mtDNA [46, 86–88, 90].
Molecular Genetic Screening
Based on the evaluation of clinical manifestations, family history, histological, and biochemical studies, screening for mutations in mtDNA- and nDNA-harboring mitochondrial genes should be carried out. First, most common point mutations in mtDNA, such as m.3243A > G, m.8344A > G, m.8993 T > C, and m.8993 T > G, can be screened using blood or urine sediment samples and PCR followed by restriction fragment length polymorphism (RFLP) [160–162]. mtDNA deletions and duplications can be identified by long-range PCR or Southern blotting techniques [6]. Quantitative changes in mtDNA can be assessed by real-time PCR using a nuclear housekeeping gene as a reference.
Mutations in the nuclear POLG1 gene are among the most frequent defects, which cause highly heterogeneous clinical phenotypes [38, 46]. Therefore, if screening for the common mtDNA mutations is negative, DNA sequencing of POLG1 should be performed in a patient with suspected mitochondrial disorder.
The most direct approach to identify molecular defect in patients with suspected mitochondrial disease is sequencing the entire mitochondrial genome or the selected nuclear genes (Fig. 24.3). Although several diagnostic laboratories use Sanger sequencing for molecular testing of mitochondrial diseases, emerging technologies such as next-generation sequencing (NGS) and high-throughput mitochondrial microarrays greatly facilitate human genomic analysis in the clinical context [19, 163–166].
Next-Generation Sequencing
The automated Singer sequencing, a first generation technique, remains the gold standard for sequencing accuracy, but it can read approximately 2 × 106 bases per day. In contrast, newer technologies referred to as NGS highly increase throughput up to 5 × 1010 bases per day. NGS relies on the unique combination of template preparation, sequencing, imaging, and data analysis (genome alignment and assembly) [163, 167–169].
Due to the great technological advancement of NGS, the cost of genome sequencing has dramatically been reduced. Several commercially available NGS instruments including Roche/454, Illumina/Solexa, Life/APG, Helicos BioSciences, Polonator Instrument, and Pacific BioSciences are in the market, and many other companies have their NGS platforms at various stages of development.
NGS-based technologies (the Illumina/Solexa platform) have been exploited in the sequencing of two acute myeloid genomes, and several candidate mutations have been identified [170, 171]. Whole-genome sequencing using NGS (the Life/APG platform) in a patient with the recessive form of Charcot- Marie-Tooth neuropathy has recently been reported [172].
Clearly, inherited cardiovascular conditions represent one of the main areas where NGS-based diagnostics can be applied [164, 173]. These new technological approaches have been applied to determine variants causing genetically heterogeneous conditions such as long QT syndrome (LQT), hypertrophic cardiomyopathy, and dilated cardiomyopathy [174–179]. Comprehensive genetic analysis based on NGS technologies is already available for 12 genes (AKAP9, ANK2, CACNA1C, CAV9, KCNE1, KCNE2, KCNH2, KCNJ2, KCNQ1, SCN4B, SCN5A, SNTA1) associated with long QT syndrome (http://www.genedx.com and http://www.sistemasgenomicos.com). Moreover, NGS has successfully been used for noninvasive prenatal detection of trisomy 21 and even the full mutational status of the fetus [180, 181]. These encouraging data confirm that NGS-based noninvasive prenatal diagnostics can be applied for screening for multiple conditions including congenital heart disease (CHD) [182, 183].
An impressive example of comprehensive NGS-based screening of a complete human genome in a defined clinical context has recently been reported [184]. Sequencing of genomic DNA has detected 2.6 million single nucleotide polymorphisms (SNPs) and 752 copy number variations (CNVs). Their integrated analysis has revealed elevated risks for myocardial infarction, type 2 diabetes, and some types of cancers. This approach has uncovered several rare variants in genes associated with sudden cardiac death, such as DSP, MYBPC3, and TMEM43. Moreover, clinically relevant information has been obtained on specific drug responses [184].
Estimation of genotype-specific responses to commonly used drugs, for example, clopidogrel, statins, and warfarin, is critically important for the efficient therapy of CVD. Genotype-guided analysis facilitates the dose predictions needed to choose individualized treatment and improve clinical outcome [185–189]. Pharmacogenetics in cardiology will continue to greatly benefit from the development of NGS-based technologies.
Mitochondrial Gene Profiling: Microarray Technologies
Great progress in another revolutionary technology, gene expression profiling using microarray technologies, has allowed high-resolution screening of the entire human genome. This technique is easier and faster than genome sequencing and enables to simultaneously analyze reliably and reproducibly the expression of tens of thousands of genes.
A microarray represents an ordered array of the nucleotide sequences (probes) placed in discrete locations on a solid support (gene chip). A sample containing labeled DNA or RNA is applied and hybridized with the probes on the chip. Scanning microscopy combined with computer analysis is used to detect and quantify the specific sequences in the analyzing sample [190–194]. The first generation of DNA arrays (macroarrays) allowed for analyses up to ∼2,000 genes, whereas modern microarrays enable to study the expression of more than 10,000 genes at once. Presently, multiple microarray platforms are commercially available such as Affymetrix, Agilent, Applied Biosystems, Illumina, and ROMA/NimbleGen. This high-throughput technology has been used for not only gene expression profiling but also detection of SNPS, evaluation of DNA methylation and microRNA status, array-comparative genome hybridizations, etc. [194].
Myriads of deregulated genes have been identified using microarrays in failing myocardium of patients with dilated cardiomyopathy [195–201]. Importantly, among deregulated genes identified in several microarray-based analyses, mitochondrial genes have been found, such as ATP5B, ATP5H, ATP5O, COX6B1, COX7A1, CYC1, NADFA10, NDUFAB1, NDUFB2, and SDHA [202].
While NGS-based approach, despite its great potential, has not yet been applied intensively to screen for mitochondrial disorders, gene profiling has been used to assess specifically the genetic status of mitochondria [19, 203]. Several human mitochondrial cDNA and oligonucleotide microarray platforms have been introduced over the past decade (Table 24.2).
Table 24.2
Major mitochondrial microarray platforms
Reference study | Microarray platform | Number of genes | Comments |
|---|---|---|---|
cDNA-based | |||
van der Westhuizen et al. [204] | – | 618 | no mtDNA genes |
Alesci et al. [15] Bai et al. [205] | hMitChip | 1,135 | 37 mtDNA genes included; hMiDas database with the dedicated software package |
Oligonucleotide-based | |||
Cizkova et al. [16] | h-MitoArray | 1,632 | |
Voss et al. [206] | huMITOchip | 2,737 | 37 mtDNA genes included |
One of the first arrays included cDNA probes for 618 nuclear genes implicated in control mitochondrial function but no probes for mtDNA genes [204]. A second cDNA microarray, called hMitChip, combined with human mitochondrial gene database, hMiDas, contained initially 501 nuclear mitochondrial genes and no mtDNA genes [16]. The latest generation of this platform, hMitChip 3.0, includes cDNA probes for 1,098 mitochondria-related nuclear genes and all 37 mtDNA genes [205]. The integration of hMitChip 3.0 and hMiDas3 with the dedicated software enables the detection of candidate genes and validation of alterations of their expression.
Two oligonucleotide-based microarray platforms, h-MitoArray and huMITOchip, have also been developed. The h-MitoArray contains 1,612 oligo probes, while the huMITOchip includes 4,825 probes [17, 206]. These probes are related to mitochondrial structure and function, apoptosis, lipid metabolism, and other cellular functions; huMITOchip includes probes for the 37 mtDNA genes. However, in contrast to the hMitChip 3.0, both h-MitoArray and huMITOchip do not have a dedicated software package.
Mitochondrial microarray technologies have been used to analyze changes in gene expression in various disorders. Dysregulation of multiple mitochondrial genes have been identified to be associated with Alzheimer’s and Parkinson’s disease [207, 208]. Alteration in expression of 119 mitochondria-related genes has been detected by the hMitChip 3.0 platform in postmortem brains of patients with posttraumatic stress disorder [209]. The hMitChip 3.0 analysis has revealed significant differences in expression of multiple mitochondrial genes between slow- and rapid-dividing melanoma cells [19]. Mitochondrial gene profiling has revealed a linkage of mtDNA variants to migraine headache and cyclic vomiting syndrome [210].
Mitochondrial Proteome
Proteomic technologies have emerged as a powerful tool, which complements NGS and gene profiling in the search for biomarkers of various mitochondrial disorders. Major modern strategies for comprehensive analysis of mitochondrial proteome have been discussed in Chap. 2. Presently, highly sensitive mass spectrometers allow the identification of thousands of different proteins from 100 μl blood samples or from ∼5,000 cells using laser capture microdissection from tissue section [211, 212]. Moreover, current proteomic techniques enable analysis of various posttranslational modifications of mitochondrial proteins, which are frequently impaired during pathological conditions [213, 214].
The mitochondrial proteomics represents one of the most actively developing fields in current mitochondrial research [213–217]. Great technological advances in this field have led to the development of several online databases of mitochondrial proteins such as MitoCarta (http://broadinstitute.org/pubs/MitoCarta/), MitoProteome (http://mitoproteome.org), MitoP2 (http://ihg.gsf.de/mitop2), MitoMiner (http://mitominer.mrc-mbu.cam.ac.uk), MitoPhenome (http://mitophenome.org), and HMPDb (http://bioinfo.nist.gov) [218–221]. However, the number of the mitochondrial proteomic analyses remains rather limited. Most studies focus on mitochondrial proteome in neurodegenerative diseases and cancer [217, 222–224]. Nevertheless, a growing number of reports have characterized specifically mitochondrial proteome in the mammalian myocardium [225–229].
To gain insight into the mechanism of HF and to assess the potential role of mitochondria in the disease, modulations in mitochondrial proteome have been examined using a number of different animal models of HF [230–235]. Major alterations in proteins implicated in fatty acid oxidation (FAO) and glycolysis as well as in OXPHOS subunits have been observed. However, the functional significance of the identified changes remains largely unclear as many of these alterations appear to be dependent on models and proteomic technologies used in the studies [236].
Cardiac mitochondrial proteomic changes in patients at different stages of HF have recently been analyzed [237]. Downregulation of proteins mediating energy generation, such as subunits of Complex I, components of TCA, and the pyruvate dehydrogenase complex, has been shown at end stage of HF, whereas their levels have been largely unchanged in “reversibly dysfunctional myocardium.” Moreover, in end-stage HF SOD2 (MnSOD) and VDAC1 have also been downregulated, suggesting reduced superoxide scavenging capacity in the human failing heart [237].
Interestingly, a number of studies have examined changes in the cardiac mitochondrial proteome associated with cardioprotection (CP) [215, 238, 239]. Several protein kinases including hexokinase, PKCε, PKCδ, and glycogen synthase kinase-3, which phosphorylate a number of mitochondrial targets, have been reported to translocate to mitochondria with CP. Accordingly, increased phosphorylation of mitochondrial proteins during preconditioning have been demonstrated [239]. Increase in S-nitrosylation of various mitochondrial proteins associated with CP has also been shown [215]. However, the role of these posttranslational modifications in CP is unclear and has to be further investigated using more sensitive proteomic techniques.
Conclusions and Future Trends
Mitochondrial disorders encompass a highly heterogeneous group of diseases characterized by not only defects in the OXPHOS system but also alterations in diverse signaling pathways, intracellular Ca2+ homeostasis, and cell survival and death. A growing number of identified disease-caused mutations in both nuclear and mitochondrial genomes make the diagnosis of mitochondrial disorders extremely challenging. The current algorithm for molecular diagnosis of patients with suspected mitochondrial disease is a complex multistep process that implicates thorough clinical testing, histochemical, and biochemical analyses followed by DNA sequencing and/or gene profiling.
The advent of NGS and global gene expression profiling technologies have enabled genome-wide screening for disease-related mutations, SNPs, and CNVs. Great technological advancements in these approaches have led to dramatic decrease in time and cost preparing them for application in clinical cardiology.
Progress in the proteomics field has not been as dramatic and has somewhat lagged behind molecular genetics. However, the development of new generation of highly sensitive mass spectrometers able to identify proteins at sub-femtomolar levels in complex biological samples as well as the design of techniques bringing quantification to the proteome will accelerate the translation of proteomic research into clinical practice [240, 241].
Recent advancements in genomics, transcriptomics, and proteomics have highlighted the critical importance of bioinformatics tools. The NIH supports bioinformatics studies directed toward management and efficient analysis of the large “metagenomic” datasets, database architecture, and integration of metagenomics with functional genomics. These efforts will accelerate the translation of genomic medicine into the clinical setting.
The advent of the “omics” era has opened up unprecedented opportunities for a system biology approach to assess the pathogenesis of CVD. Despite the great potential of this systemic approach, it is not as extensively used in clinical cardiology as, for example, in clinical oncology [242]. Nevertheless, these revolutionizing technologies have started to be applied to diagnosis of CVD, although their use for the diagnosis of mitochondria-related cardiac diseases is yet lagging behind and remains rather limited. Currently, DNA sequence analysis of only approximately 150 genes related to mitochondrial disorders is clinically available leaving a majority of patients with suspected mitochondrial disorders without a definitive diagnosis [15].
Finally, in contrast to significant progress in deciphering the molecular mechanisms underlying mitochondrial dysfunction, there is no satisfactory mitochondrial gene therapy protocol thus far. While experimental therapeutic strategies have recently been suggested, there are still significant technical difficulties to overcome before they can be used for the treatment of patients with mitochondria-related cardiac diseases [243, 244].
The convergence of mitochondrial research and high-throughput genomic, transcriptomic, proteomic, and bioinformatic technologies will improve our understanding of the role of this organelle in the heart and will ultimately lead to more individualized targeted therapies for mitochondria-related CVD.
Summary
The clinical study by Luft et al. [3] showing mitochondrial abnormalities in muscle of a patient with severe hypermetabolism of nonthyroid origin has introduced the concept of mitochondrial disorders and mitochondrial medicine. However, only more than two decades later, it was found that mutations in mtDNA were associated with human diseases, myopathies, and optic neuropathy, and the molecular era of mitochondrial medicine was originated.
Mitochondrial disorders are genetically heterogeneous and display very variable clinical features. Recent studies estimate that ∼1 in 5,000 children will develop a mitochondrial disorder and ∼1 in 200 adult individuals carry mtDNA mutations and can transmit mitochondrial diseases.
mtDNA is characterized by significantly higher (10- to 20-fold) mutation rate compared to its nuclear counterpart. To date more than 300 pathogenic mtDNA mutations, including point mutations and rearrangements, mainly large-scale (1.3–8 kb) deletions spanning several genes, have been reported (http://www.mitomap.org).
One of the most common mtDNA point mutation is the m.3243A >G mutation in tRNALeu(UUR), which causes MELAS syndrome or diabetes, hearing loss, and retinopathy. Another common point mutation, m.8344A >G in tRNALys, causes MERRF syndrome. The genes encoding tRNALeu(UUR) and tRNALys represent mutation hot spots within which several disease-causing mutations have been identified.
The m.1494 T >C and m.1555A >G mutations are point mutations in the mitochondrial 12S rRNA gene associated with deafness. Examples of disease-causing mutations in the genes encoding proteins include m.8993 T >C and m.8993 T >G in the ATP6 gene, which cause maternally inherited Leigh syndrome (MILS) and neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome, respectively, and m.3460 G >A, m.11778 G >A, and m.14484 T >C in the ND1, ND4, and ND6 genes, respectively, which cause Leber hereditary optic neuropathy (LHON).< div class='tao-gold-member'>Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


