(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
All multicellular organisms develop during their evolution highly regulated and interconnected pathways of cell death. While cell death has initially been considered as an accidental process, it is now recognized that many aspects of cell death are tightly regulated. Cell death can be classified into three types: apoptosis, necrosis, and autophagy. A variety of stimuli can initiate all three types of cell-death pathways in the myocardium. This complex, highly interconnected network contributes to the pathogenesis of various cardiovascular disorders including ischemia/reperfusion (I/R) injury, myocardial infarction, heart failure, dysrhythmias, and atherosclerosis. Mitochondria are central mediators of cardiomyocyte survival and death. Two major apoptotic pathways, mitochondria (intrinsic) and death-receptor-mediated (extrinsic), converge at the mitochondria inducing release mitochondrial apoptogens to initiate caspase cascade and eventually degradation of the doomed cardiomyocyte. Activation of death receptors can initiate not only extrinsic apoptotic pathway but also necrosis. Mitochondria permeability transition pore (MPTP) opening is an essential mechanism underlying necrosis, which is mediated by increased oxidative stress, elevated Ca2+ and phosphate concentrations, and adenine nucleotide depletion. Autophagy, which is characterized by the massive formation of lysosomal-derived vesicles, containing degenerating cytoplasmic contents, is primarily a survival response to nutrient deprivation. mTOR and Beclin-1 are two main regulators of autophagy. Selective form of autophagy, mitophagy, is also a protective mechanism to eliminate damaged mitochondria and thereby to attenuate mitochondria-mediated apoptosis and necrosis in the myocardium. Further insight into the molecular mechanisms underlying intricate network of cell-death pathways will increase the efficiency and repertoire of therapeutic interventions in cardiovascular disease.
Introduction
All multicellular organisms develop during evolution the highly regulated and interconnected pathways of cell death. Although cell death has initially been considered as an accidental uncontrolled process, it is now well recognized that many aspects of cell death are genetically programmed and therefore actively and tightly regulated. Currently, cell death can be classified into three types: apoptosis, necrosis, and autophagy (Fig. 11.1) [1–4]. While this classification is based mainly on morphological criteria, recent progress in research has provided insights into the molecular distinctions between different types of cell death.
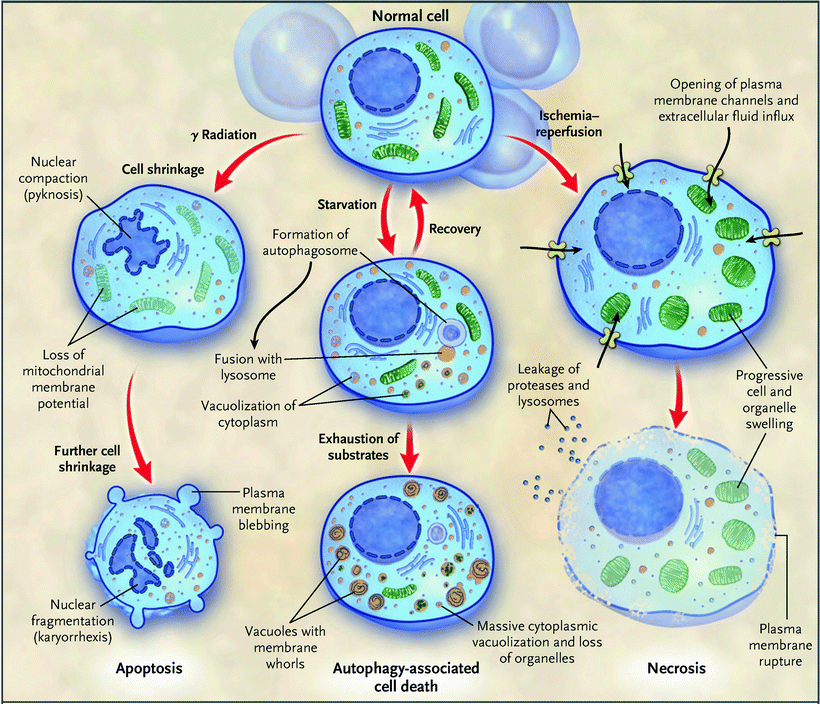
Fig. 11.1
Schematic representation of three modes of cell death. Morphological features of the three major modes of cell death—apoptosis, autophagy, and necrosis—are schematically depicted. Different types of injury can induce predominantly a particular pathway of cell death. Intensive communications between different pathways of cell death are not shown (Adapted from Hotchkiss et al. [3] with permission of the Massachusetts Medical Society)
Mitochondria play a central role not only in maintaining the life of cardiomyocytes by providing ATP through oxidative phosphorylation (OXPHOS) but also in regulation of cell death, responding to various stress signals. The recognition that mitochondria are key regulators of the cardiomyocyte’s decision to live or die has revolutionized cardiovascular research. Currently, it is widely recognized that mitochondrial dysfunction contributes to the pathogenesis of various cardiovascular diseases (CVD) including ischemia/reperfusion (I/R), cardiomyopathy, myocarditis, heart failure (HF), and atherosclerosis.
In this chapter, we discuss recent progress in our understanding of the molecular mechanisms of apoptosis, necrosis and autophagy, and role of cardiac mitochondria in these processes.
Apoptosis
The term “apoptosis” (from an ancient Greek word describing falling leaves) was introduced by Kerr, Wyllie, and Currie in 1972 to designate a regulated cell death process distinct from that of “necrosis,” which was considered an accidental and passive type of cell death [5]. One of the distinctions between apoptosis and necrosis is the different dynamics of the loss of plasma membrane integrity. In apoptosis, plasma membrane maintains its integrity until the late stages of the process, while in necrosis, plasma membrane rupture and resultant swelling of organelles and loss of intracellular contents are early events [2, 3, 6, 7]. Apoptosis is characterized by several morphological features including cell rounding-up, reduction of cellular volume (pyknosis), chromatin condensation, nuclear fragmentation (karyorrhexis), and plasma membrane blebbing.
An essential process of apoptosis is activation of a class of specific proteases, caspases (cysteinyl aspartyl proteases) [8, 9]. They are synthesized as inactive procaspases and can be subdivided into upstream procaspases 2, 8, 9, and 10 and downstream procaspases 3, 6, and 7. Upstream procaspases are usually monomeric proteins that in response to apoptotic stimuli are activated by dimerization followed by autocleavage [10]. The resulting activated caspases cleave downstream caspases 3 and 7, which degrade myriads of target proteins and eventually dismantle the doomed cardiomyocyte
Two distinct but interlinked pathways mediate apoptosis: the intrinsic or mitochondrial pathway and the extrinsic or death-receptor pathway (Fig. 11.2).
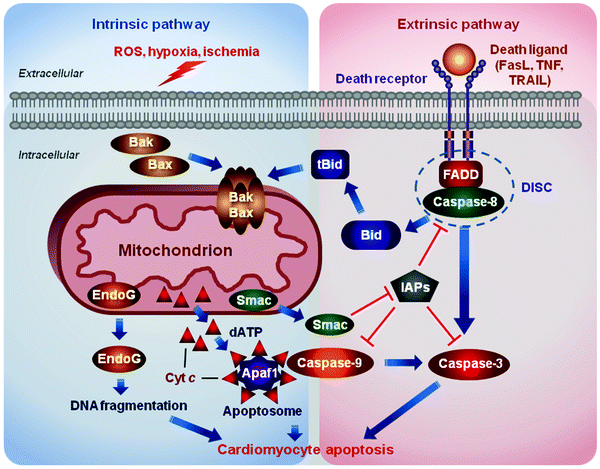
Fig. 11.2
Two major apoptosis pathways. The intrinsic or mitochondrial pathway is induced by diverse stress stimuli, including growth factor deprivation, oxidative and genotoxic stress, hypoxia, and various toxins. These signals are transduced by the B-cell lymphoma 2 protein (Bcl-2) family proteins to mitochondria and sarcoplasmic reticulum (not shown). Pro-apoptotic Bcl-2-associated X protein (Bax)/Bcl-2 homologous antagonist/killer (Bak) induces the release of mitochondrial apoptogens, such as cytochrome c (Cyt c), endonuclease G (EndoG), and Smac/DIABLO (Smac), into the cytosol. Released Cyt c along with deoxyadenosine triphosphate (dATP) binds to apoptotic protease activating factor 1 (Apaf1) to form a heptameric protein complex, apoptosome. The apoptosome promotes cleavage and activation of procaspase 9. Activated caspase 9 in turn cleaves and activates numerous downstream procaspases, proteases, and DNases resulting eventually in the cell demolition. The extrinsic or death-receptor pathway is triggered by binding of death ligands [e.g., Fas ligand (FasL), tumor necrosis factor (TNF), or TNF-related apoptosis-inducing ligand (TRAIL)] to their cognate receptors. Engaged death receptors recruit the adaptor protein “Fas-associated protein with death domain” (FADD), which in turn recruits procaspase 8, assembling the death-inducing signaling complex (DISC). DISC formation triggers activation of caspase 8, which in turn cleaves and activates downstream effector procaspases. Caspase 8 can also cleave BH3 interacting-domain death agonist (Bid) to generate its truncated form (tBid), which translocates to mitochondria to activate pro-apoptotic Bax/Bak. Inhibitors of apoptosis proteins (IAPs) block caspase activation, while Smac/DIABLO, released upon mitochondrial permeabilization, suppresses the action of IAPs. ROS reactive oxygen species
Intrinsic (Mitochondrial) Pathway
The mitochondria pathway is induced by various stress stimuli, including growth factor deprivation, oxidative and genotoxic stress, hypoxia, and various toxins. These signals are transduced by the Bcl-2 family proteins to mitochondria, leading to release of mitochondrial apoptotic proteins, and to sarcoplasmic reticulum (SR), resulting in release of Ca2+.
The BCL-2 (B-cell lymphoma-2) gene was originally identified as the gene, deregulated by the t(14;18) chromosome translocation in B-cell follicular lymphomas, which was able to inhibit apoptosis [11, 12]. The Bcl-2 family proteins are critical regulators of the mitochondrial apoptotic pathway. Members of the Bcl-2 protein family can be divided into three subtypes: the pro-apoptotic BH3-only proteins, the anti-apoptotic, pro-survival Bcl-2-like proteins, and the pro-apoptotic, pore-forming Bax and Bak proteins [13, 14].
The Bcl-2 proteins are small, globular proteins, which are characterized by the presence of the Bcl-2 homology (BH) domains [13, 14]. The anti-apoptotic Bcl-2, Bcl-XL, Bcl-W, Mcl-1, A1, and Boo contain four BH domains, BH1–BH4 (Fig. 11.3). The pro-apoptotic Bax (Bcl-2-associated X protein) and Bak (Bcl-2 homologous antagonist/killer) proteins also contain four BH domains. The BH3-only proteins (Bim, Bid, Puma, Bad, Noxa, Bmf, Bik, and Hrk), containing only the BH3 domain, are able to bind and inhibit the pro-survival Bcl-2 proteins, releasing thereby the pro-apoptotic Bax and/or Bak (Bax/Bak) and perhaps Bok (Bcl-2-related ovarian killer protein).
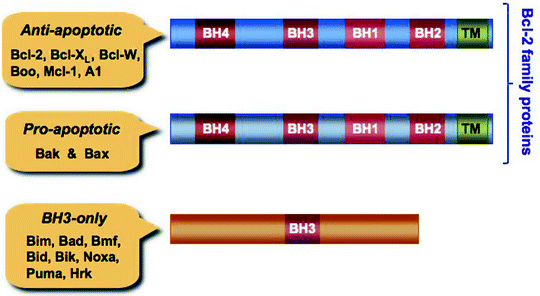
Fig. 11.3
The Bcl-2 family proteins and BH-3 only proteins. Anti-apoptotic B-cell lymphoma 2 protein (Bcl-2) family members and pro-apoptotic, pore-forming proteins, Bcl-2 homologous antagonist/killer (Bak) and Bcl-2-associated X protein (Bax), contain four Bcl-2 homology (BH) domains, while the pro-apoptotic BH3-only proteins contain only one BH3 domain. The BH3 domain of the BH3-only proteins is involved in their interaction with Bcl-2 family proteins. Predicted C-terminal transmembrane (TM) domains are also shown
In healthy cells, most anti-apoptotic Bcl-2 proteins reside constitutively in the mitochondrial outer membrane (MOM) or in the SR, where they bind and inhibit the pro-apoptotic Bcl-2 proteins. Bax is localized to the cytosol as an inactive monomer, while Bak can be found predominantly in the cytosol with a minor fraction loosely attached to the MOM (Fig. 11.4). In response to apoptotic signals, Bax and Bak undergo conformational activation leading to their recruitment to the MOM [15–18]. At the mitochondria, these multi-domain proteins form homo- and hetero-oligomers, which appear as foci, promoting MOM permeabilization [19, 20].
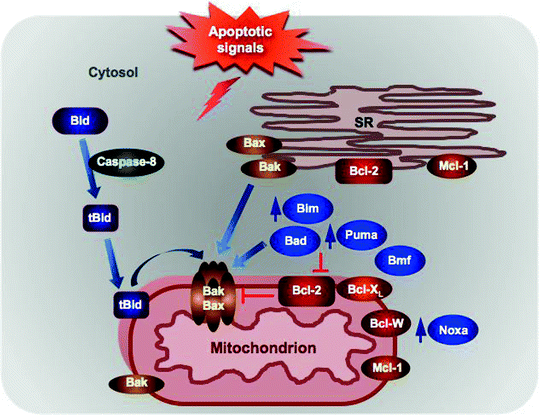
Fig. 11.4
Localization of the Bcl-2 family proteins and BH-3 only proteins. Anti-apoptotic Bcl-2 resides in the SR and in the mitochondrial outer membrane (MOM), where it binds and inhibits the pro-apoptotic Bax/Bak. Bax is localized to the cytosol as an inactive monomer, while Bak can be found predominantly in the cytosol with a minor fraction loosely attached to the MOM. In response to apoptotic signals, Bax translocates to the MOM to form homo- and heterodimers with Bak and promotes the mitochondrial permeabilization. Bid is cleaved by caspase 8, and resultant tBid translocates to mitochondria where it activates Bax/Bak. Expression of some BH3-only proteins, such as Bim, Noxa, and Puma, is induced in response to apoptotic stimuli, while others, such as Bad, Bid, and Bim, are posttranslationally modified leading to their recruitment to mitochondria. At the mitochondria, they can either activate directly the pro-apoptotic Bax/Bak or inhibit the anti-apoptotic Bcl-2 proteins. See text for details
Apoptotic stimuli induce transcription of the BH3-only proteins (Bim, Noxa, and Puma), decrease in turnover of Noxa, induce posttranslational modification such as phosphorylation or cleavage (Bad and Bid, respectively) and/or a change in binding partner and intracellular localization (Bim) [21–25]. All of these changes facilitate translocation of the BH3-only proteins to mitochondria where they interact with Bax/Bak. Although the precise mechanism of action of BH3-only proteins is currently unknown, it has been suggested that they can bind directly to Bax/Bak contributing to their oligomerization, alternatively they can displace the pro-survival Bcl-2 or Bcl-XL from Bax/Bak activating thereby the latter indirectly (Fig. 11.5) [26–28]. The binding sites on Bax and Bak are also subject of debates and intensive investigations [29–33].
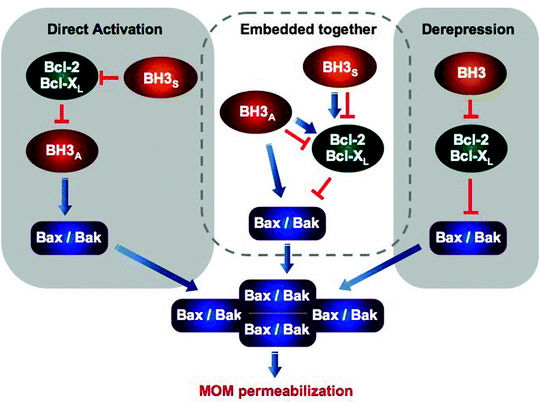
Fig. 11.5
Three models of apoptosis regulation by the Bcl-2 family proteins and BH-3 only proteins. Two contrasting models, the direct activation and derepression, postulate different functional states of Bax/Bak and therefore different mechanism by which Bcl-2/Bcl-XL attenuate MOM permeabilization. According to the direct activation model, Bax/Bak are inactive and must be activated by BH3 activators (BH3A), while Bcl-2/Bcl-XL sequester the activators inhibiting thereby apoptosis. BH3 sensitizers (BH3S) can displace Bcl-2/Bcl-XL from BH3A contributing to Bax/Bak activation indirectly. The derepression model postulates that Bax/Bak are constitutively active and Bcl-2/Bcl-XL must continuously bind and inhibit Bax/Bak to prevent MOM permeabilization and apoptosis. Finally, recently proposed “embedded together” model states that Bcl-2 proteins display both functions postulated by the other two models and BH3 proteins can not only inhibit but also activate the anti-apoptotic Bcl-2 proteins facilitating their targeting to MOM. Most of these interactions occur within mitochondrial membrane; however, in this simplified scheme, its important role is not shown
Conversion of Bax/Bak into oligomerized activated form, resulting in generation of the “apoptotic pore” in the MOM, represents the point of no return in mitochondrial apoptotic pathway. The paramount role of Bax/Bak in mediating apoptosis has been highlighted by the high resistance to apoptosis of bax−/−bak−/− double-knockout cells [34]. In the myocardium, activation and recruitment of Bax to the MOM have been demonstrated during oxidative stress (OS) and I/R [35–37]. Interestingly, Bax deficiency in mice has resulted in reduced myocardial infarct (MI) size and decreased number of damaged mitochondria compared to wild-type control animals [38]. Furthermore, elevated levels of Bak have been demonstrated in patients with dilated cardiomyopathy; however, its role in cardiac disorder remains to be determined [39].
Permeabilization of the MOM promotes the release of multiple apoptogens (e.g., cytochrome c, Smac/DIBLO, endonuclease G [endoG]) from the mitochondrial intermembrane space into the cytosol (Figure 11.2) [40–42]. Released cytochrome c, which is normally a component of ETC, along with the cytosolic deoxyadenosine triphosphate dATP binds to the apoptotic protease activating factor 1 (Apaf1). These interactions induce conformational changes in Apaf1 leading to its oligomerization and formation of a heptameric protein complex, apoptosome. The apoptosome can bind procaspase 9 promoting its cleavage and activation. Activated caspase 9 in turn cleaves and activates numerous downstream procaspases, proteases, and DNases resulting eventually in the cell demolition [42, 43].
Several proteins belonging to the inhibitor of apoptosis (IAP) family can bind and inhibit caspases. For example, X-linked inhibitor of apoptosis protein (XIAP) inhibits already activated caspases 3 and 7 and also binds procaspase 9 preventing its activation within the apoptosome [44–47]. In addition, IAPs due to their E3 ubiquitin ligase activity can promote proteasomal destruction of caspases in response to apoptotic stimuli [48, 49].
Other released mitochondrial pro-apoptotic factors, Smac/DIABLO (second mitochondria-derived activator of caspase/direct IAP-binding protein with low PI) and Omi/high temperature requirement protein A2 (Omi/HtrA2), can bind the IAPs thereby releasing the caspases [41, 50–52]. Moreover, Omi/HtrA2 is a serine protease, which can degrade XIAP facilitating pro-apoptotic caspase activity [53]. Finally, endoG, when released from the mitochondrial intermembrane space, catalyzes DNA fragmentation contributing to the cell destruction.
Extrinsic Pathway
This apoptotic pathway, also known as the death-receptor pathway, is activated by the binding of death ligands, such as Fas ligand (FasL), tumor necrosis factor (TNF), or TNF-related apoptosis-inducing ligand (TRAIL), to their cognate cell surface death domain-containing receptors, commonly known as death receptors (Fig. 11.2) [54]. Death receptors belong to the large TNF receptor family characterized by the presence of conserved intracellular death domains (DD), which are essential for the initiation of apoptotic response [55]. Fas (CD95/Apo1), TNF-R1 (CD120a/p55), DR3 (TRAMP), DR4 (TRAIL-R1), and DR5 (TRAIL-R2/Apo2/TRICK2/KILLER) are the most well-characterized death receptors. They share common activation mechanism: binding of specific ligands to their respective receptors induces receptor homo-trimerization, which triggers the recruitment of cytosolic adaptor and effector proteins [56]. Fas, DR4, and DR5 recruit the Fas-associated death domain (FADD) protein and mediate primarily apoptosis, while TNF-R1 and DR3 recruit TRADD and promote mainly pro-inflammatory response. FADD associated with activated receptor in turn recruits procaspase 8 or 10 to assemble the multiprotein death-inducing signaling complex (DISC) [57]. DISC formation induces conformational changes in its subunits resulting in activation of caspase 8 or 10, which in turn cleave and activate downstream effector procaspases, primarily procaspases 3, 6, and 7 [58, 59].
Although the DISC-mediated direct activation of effector procaspases may be sufficient in some cells, so-called type I cells, in most cells (type II cells) activation of the BH3-only protein Bid is required for the efficient apoptotic response. The DISC-activated caspase 8 cleaves Bid into a pro-apoptotically active 15-kDa truncated form, tBid [60, 61]. Other caspases such as caspases 3 and 10 are also capable of cleaving Bid [62–64]. The resultant tBid translocates to the mitochondria and induces MOM permeabilization leading to the release of apoptogens and to the mitochondria-dependent apoptosis as discussed above. It has recently been demonstrated that a novel tBid-interacting mitochondrial carrier homologue 2/Met-induced mitochondrial protein (MTCH2/MIMP) facilitates the recruitment of tBid to mitochondria resulting in MOM permeabilization [65]. Thus, tBid not only facilitates the extrinsic pathway but also provides the link between the death-receptor and mitochondrial apoptotic pathways.
Necrosis
“Necrotic cell death” or “necrosis” (from a Greek word “necros” for corpse) is characterized morphologically by cell and organelle swelling (oncosis), early plasma membrane rupture, and resultant loss of intracellular contents [7, 66]. For a long time, this type of cell death has been viewed, in contrast to apoptosis, as a purely accidental uncontrolled process. However, emerging evidence suggests that many aspects of necrosis could be finely regulated. Some authors have proposed the term “necroptosis” to describe regulated necrotic cell death [67, 68]. Although it is becoming evident that multiple signaling pathways are involved in necrosis, the precise molecular mechanisms of this process have only just begun to emerge.
Over the past decade, it has been demonstrated that activation of death receptors can initiate not only extrinsic apoptotic pathway but also necrosis. TNF signaling represents the most extensively characterized example [69–71]. The binding of TNF-α to TNF-R1 triggers the assembly of the membrane-proximal complex I that includes also TNF-R-associated death domain (TRADD), receptor-interacting protein 1 (the serine/threonine kinase RIP1), cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1 and 2), and TNF-R-associated factor 2 and 5 (TRAF2 and 5) (Fig. 11.6) [72]. cIAPs are E3 ubiquitin ligases, which catalyze Lys63 polyubiquitination of RIP1 and TRAF2 [73–75]. Polyubiquitinated RIP1 and TRAF2 bind TGF-β-activated kinase 1 (TAK1) along with TAK1-binding proteins 2 and 3 (TAB2 and 3). The formed TAK1–TAB2/3 complex mediates the canonical nuclear factor-κB (NF-κB) activation pathway, which transactivates multiple pro-survival genes [76]. However, complex I has been reported to mediate also the recruitment of the ROS-generating NOX1 NADPH oxidase to the plasma membrane contributing thereby to necrosis [77, 78]. Thus, depending on cell type and/or stimuli, complex I can promote either pro-survival (via NF-κB pathway) or pro-death (via NOX1 engagement) responses.
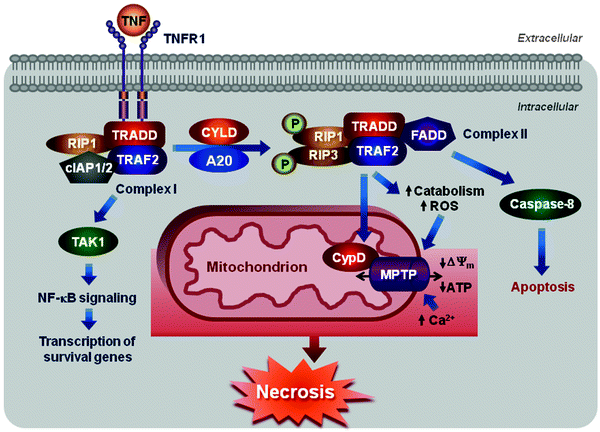
Fig. 11.6
Scheme of necrotic cell death. Engagement of TNFR1 triggers the assembly of the membrane-proximal complex I composed of TNFR1, TRADD, RIP1, cIAP1/2, and TRAF2/5. Complex I can mediate through TAK1 recruitment activation of NF-κB pathway leading to upregulation of survival genes. Depending on context, complex I can also undergo endocytosis resulting in dissociation of TNFR1, deubiquitination of RIP1 by CYLD and A20, and recruitment of FADD and procaspase 8 to form complex II. Within complex II, procaspase 8 is activated and can promote apoptosis by activating the caspase cascade. Caspase 8 can also cleave and inactivate RIP1 and 3 inhibiting thereby either necrosis or pro-survival NF-κB-mediated pathway. However, if caspase 8 is inhibited, RIP1 can recruit RIP3 to the complex, where they are phosphorylated, and complex II promotes MPTP opening via its subunit CypD activating eventually necrosis. See text for details
Internalization of complex I results in dissociation of TNF-R1, deubiquitination of RIP1 by cylindromatosis (CYLD), and recruitment of Fas-associated protein with a death domain (FADD) and procaspase 8. Resultant multiprotein signaling complex is known as complex II [72, 79, 80]. Within complex II, procaspase 8 is activated and can promote apoptosis by activating the caspase cascade. Caspase 8 can also cleave and inactivate RIP1 and 3 inhibiting thereby either necrosis or pro-survival NF-κB-mediated pathway [71, 81–83]. However, if caspase 8 is inhibited pharmacologically or deleted, depleted genetically, RIP1 can recruit RIP3 to the complex, where they are phosphorylated, and complex II promotes necrosis [83, 84]. The RIP1–RIP3 interactions and their kinase activities appear to be critical for this pathway, although their downstream targets are largely unknown.
The opening of the mitochondrial permeability transition pore (MPTP) is another essential mechanism underlying necrosis. MPTP opening is induced by excessive OS and elevated Ca2+ and phosphate concentrations and adenine nucleotide depletion. This leads to the dissipation of the Δψm, inhibition of ATP synthesis, increased ROS generation, and eventually in swelling and rupture of the mitochondrion, a hallmark of necrosis. Mitochondrial damage leads to the release of the described above pro-apoptotic proteins; however, necrotic mitochondrial membrane rupture appears to differ from the mitochondrial membrane permeabilization during apoptosis.
Although molecular structure and composition of the MPTP remains to be determined, cyclophilin D (CYPD) is a critical component of the MPTP, which regulates its opening [85–87]. Cells deficient for CYPD are resistant to cell death triggered by Ca2+ overload and OS, while transgenic CYPD overexpression leads to spontaneous mitochondrial swelling and necrotic cell death [85, 88, 89]. Peptidyl-prolyl cis–trans isomerase activity of CYPD is essential for its ability to regulate MPTP and required for necrosis.
Poly-ADP-ribose polymerase (PARP), Ca2+, and non-caspase proteases, calpains and cathepsins, represent other important mediators of necrotic cell death [90–96]. PARP catalyzes the repair of DNA strand breaks. Since this reaction is ATP- and NAD-consuming, PARP-mediated DNA repair can deplete intracellular ATP and NAD levels thereby shifting cell death from apoptotic to necrotic mode. Importantly, PARP cleavage upon onset of apoptosis, which serves as an early diagnostic marker of this process, inactivates the enzyme and thereby preserves intracellular ATP [97]. In addition to ATP depletion, PARP can promote release of apoptosis-inducing factor (AIF) from the mitochondrial intermembrane space caused by DNA damage [98, 99]. Released AIF translocates into the nucleus where it mediates large-scale DNA fragmentation, which in turn further activates PARP, establishing a vicious cycle [100]. Accordingly, pharmacological and genetic inhibition of PARP results in decreased necrosis during I/R injury [90, 101].
Increased intracellular Ca2+ concentration is an essential characteristic of necrosis. Influx of extracellular Ca2+ mediated by the voltage-sensitive L-type channels, acid-sensing ion channels, and transient receptor potential channels can induce Ca2+ release from the ER/SR stores [92, 93, 102, 103]. These events contribute to mitochondrial Ca2+ overload leading eventually to necrotic cell death. In addition, Ca2+ overload activates multiple enzymes, such as phospholipases, nitric oxide synthases, lysosomal hydrolases, and proteases, which contribute to the initiation of necrosis.
Ca2+ induces non-lysosomal, non-caspase cysteine proteases, calpains, ubiquitously expressed in mammalian cells [104]. Calpains can trigger apoptosis by cleaving and activating the pro-apoptotic Bid and Bax [105, 106]. However, calpains can also cleave upstream and downstream procaspases into inactive fragments inhibiting thereby apoptotic and activating necrotic cell death [107, 108].
Lysosomal cathepsins are another group of proteases, predominantly endopeptidases, which can be released from lysosomes in response to death stimuli [96, 109, 110]. Cathepsin D is implicated in cyclosporine-induced necrosis in human umbilical artery cells; however, the exact mechanism of this pathway as well as the role of lysosomes in necrosis remains to be determined [111].
Autophagy
“Autophagy” or “autophagic cell death” (from the Greek “phagy”—to eat, and “auto”—oneself) is morphologically characterized by the massive formation of single- or double-membrane lysosomal-derived vesicles, which contain degenerating cytoplasmic particles, organelles, and protein aggregates (Fig. 11.1) [112–115]. This catabolic process occurs typically in the absence of chromatin condensation. Autophagy is primarily a survival response to nutrient deprivation to recycle intracellular components and provide the cell with metabolites. [116, 117] It is also a protective mechanism to eliminate damaged cellular organelles and toxic misfolded proteins [118–120].
Three main types of autophagy have been described: microautophagy, macroautophagy, and chaperone-mediated autophagy [113, 116]. Microautophagy is mediated by an invagination of the lysosomal membrane, which engulf the cytosolic contents and deliver them to degradation. In macroautophagy (herein referred to as autophagy), the newly formed double-membrane vesicle (autophagosome) surrounds and sequesters cytoplasmic organelles and proteins. The autophagosome then fuses with the lysosome, forming autolysosome, where the cargo and inner layer of the double autophagosomal membrane are degraded by lysosomal enzymes. In chaperone-mediated autophagy, heat shock proteins bind to target proteins and deliver them to lysosomes for degradation.
Mammalian target of rapamycin (mTOR) and Beclin-1 are two main regulators of autophagy (Fig. 11.7). mTOR, a serine–threonine protein kinase, is the central sensor of nutrient status, which is activated upon fed conditions to stimulate protein synthesis and inhibit autophagy via phosphorylation and inactivation of autophagy-related proteins (Atg) [121, 122]. AMP-activated protein kinase (AMPK) can inhibit mTOR and thereby override mTOR-mediated suppression of autophagy. In response to starvation, class I phosphatidylinositol-3-kinase (PI3K) signaling promotes mTOR inhibition resulting in suppression of protein synthesis and activation of autophagy [123].
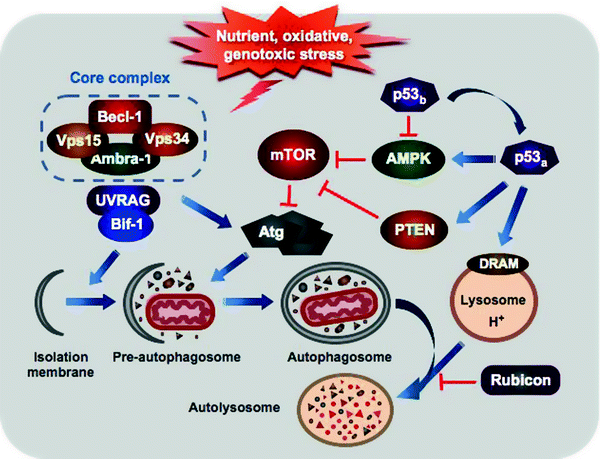
Fig. 11.7
Major regulatory and morphologic steps of autophagy. Various sublethal stress stimuli induce autophagy through activation of the class III PI3K (Vps34) or inhibition of mTOR. Vps34 along with Beclin-1, Vps15, and Ambra1 forms the core complex, which is further activated by the interaction with UVRAG and Bif-1 to initiate vesicle nucleation. A complex cascade of the Atg-ubiquitin conjugation events mediates elongation of the pre-autophagosome and attachment to damaged organelles and protein aggregates. Maturation of the pre-autophagosome followed by its closure generates the double-membrane autophagosome. The mature autophagosome fuses with lysosome delivering its contents for degradation by the lysosomal enzymes. Rubicon can attenuate autophagosome fusion with lysosome. p53 regulates autophagy in dual fashion: basal p53 levels (p53b) inhibit AMPK relieving mTOR-mediated suppression of autophagy, while stress-activated p53 (p53a) induces autophagy through both AMPK activation and transcriptional upregulation of PTEN and DRAM. See text for details
The p53 tumor suppressor, “guardian of cell genome,” not only regulates cell cycle, DNA repair, and apoptosis but also governs autophagy. p53 can control autophagy in dual fashion: basal p53 levels inhibit AMPK relieving mTOR-mediated suppression of autophagy, while p53 activation induces autophagy through AMPK activation and transcriptional upregulation of PTEN, DRAM, and TSC1 [124–126]. Importantly, activation of p53 by distinct stress stimuli induces apoptosis through engagement of Puma and Noxa [117, 127].
Human BH3-only protein Beclin-1 is a functional homolog of yeast Atg6/vacuolar protein sorting 30 (Vps30) [128, 129]. In association with the class III PI3K (PI3K3C)/Vps34, the regulatory myristoylated kinase Vps15, and possibly Ambra1, Beclin-1 forms the core complex [130–133]. Binding of Atg14 and possibly UV irradiation resistance-associated gene (UVRAG) and Bif-1 activates the core complex to initiate vesicle nucleation. [133–135] On the other hand, it has recently been reported that Atg14 and UVRAG appear to be present in mutually exclusive Beclin-1–Vps34–Vps15 complexes [136]. Moreover, UVRAG is also involved in autophagosome maturation and endocytic trafficking [137]. While Atg14, UVRAG, and Bif-1 activate autophagosome formation, recently identified component of Beclin-1–Vps34–Vps15 complexes, Rubicon (RUN domain and cysteine-rich domain containing, Beclin-1-interacting protein), negatively regulates this process [133, 138].
A complex cascade of the Atg-ubiquitin conjugation reactions mediates elongation of the pre-autophagosome and attachment to damaged organelles and protein aggregates. Maturation of the pre-autophagosome followed by its closure generates the double-membrane autophagosome [114, 139]. The mature autophagosome fuses with lysosome delivering its contents, such as pro-apoptotic mitochondria, for destruction by the lysosomal enzymes.
While autophagy has initially been viewed as a nonselective process for degrading damaged organelles and protein aggregates, it becomes clear that molecular mechanisms exist for the selective recruitment of the cargo to autophagosomes. The term “mitophagy” has been introduced to describe selective autophagic removal of damaged mitochondria [140]. Recent studies suggest a number of factors such as Atg32, Parkin, PTEN-induced putative kinase 1 (PINK1), BNip3, and Nix (also called BNip3L), which confer selectivity during mitophagy [141–147]. However, the mechanisms by which these factors mediate this process remain to be determined. Other cargo-specific autophagic pathways are ER-phagy, pexophagy (peroxisome degradation), ribophagy (ribosome degradation), and xenophagy (intracellular pathogens elimination).
Initially, Beclin-1 was identified as a protein, which interacts with anti-apoptotic Bcl-2 proteins, such as Bcl-2 and Bcl-XL [148]. This interaction, mediated by its BH3-only domain and the BH3 receptor domain of Bcl-2 and Bcl-XL, sequesters Beclin-1 from the Beclin-1–Vps34–Vps15 core complex and inhibits thereby autophagy induced by nutrient depletion [129, 149]. Consistently, mutations within these domains disrupt the Beclin-1–Bcl-2/Bcl-XL complex leading to the induction of autophagy. In contrast, the Beclin-1–Bcl-2 interaction does not affect the anti-apoptotic function of Bcl-2 [150].
It has recently been demonstrated that both components of the autophagy-promoting core complex, Beclin-1 and PI3K3C/Vps34, can be cleaved by caspases 3, 7, and 8 in response to activation of both mitochondrial and death-receptor apoptotic pathways [151, 152]. Moreover, in contrast to full-length Beclin-1, its N- and C-terminal cleavage fragments fail to induce autophagy [153]. More importantly, the C-terminal fragment of Beclin-1 acquires a new function facilitating mitochondria-mediated apoptosis. Thus, Bcl-2/Bcl-XL–Beclin-1 interactions represent not only a key mechanism to switch on or off autophagy but also an important link between different cell-death pathways [154, 155].
FLIP (Flice inhibitory protein), an inhibitor of death-receptor apoptotic pathway, is also involved in cross talk between autophagy and apoptosis. It suppresses autophagy through a direct interaction with Atg proteins essential for autophagosome elongation [156].
Cell Death in the Pathogenesis of Myocardial Disorders
All three types of cell death, apoptosis, necrosis, and autophagy, occur in the heart during I/R, MI, and HF. Growing evidence suggests that multiple aspects of these cell-death pathways are tightly controlled. Cardiac mitochondria play critical roles in these processes.
Permanent coronary occlusion and I/R induce myocardial cell death in the ischemic zone, which is mediated by apoptosis, necrosis, and autophagy [88, 89, 157–161]. Although it is difficult to estimate the prevalence and timing of the various forms of cell death, it appears that cardiomyocyte apoptosis peaks at 4.5 h, while necrosis at 24 h after permanent coronary occlusion [158]. It has been reported that I/R accelerates the apoptosis timing in the myocardium compared to occlusion [162].
Both the mitochondrial and death-receptor apoptotic pathways contribute to the pathogenesis of MI. Deletion of pro-apoptotic Bax or myocardium-specific overexpression of anti-apoptotic Bcl-2 attenuates cardiomyocyte apoptosis and protects against myocardial I/R injury [38, 163, 164]. Fas signaling pathway mediates myocardial apoptosis during I/R. Deficiency in Fas reduces infarct size, while deletion of both receptors TNF-R1 and 2 exacerbates infarction size following permanent coronary occlusion [165, 166].
Cleavage and activation of Bid by caspase 8 or calpain, the event linking the intrinsic and extrinsic apoptotic pathways, has been demonstrated during cardiac I/R [105, 167]. Consistently, mice deficient in Bid or Puma have reduced cardiomyocyte apoptosis, infarct size, and improved cardiac function during I/R [168].
Apoptosis repressor with caspase recruitment domain (ARC), an inhibitor of apoptosis highly expressed in skeletal and cardiac myocytes, attenuates both intrinsic and extrinsic pathways [169–171]. Transgenic overexpression of ARC protects the heart from I/R injury [172].
Multiple events during I/R, such as hypoxia, intracellular acidosis, Ca2+ overload, and increase in ROS generation, contribute to MPTP opening leading eventually to necrotic cardiomyocyte death. Deficiency in CYPD, an essential regulator of MPTP opening, results in attenuated necrosis and reduced infarction size following I/R [88, 89]. Inhibition of another important player in necrotic pathway, RIP1, by small molecule necrostatin-1 also reduces I/R-induced infarct size [173–175]. Moreover, the cardioprotective effect of necrostatin-1 requires the presence of CYPD suggesting a connection between RIP1 signaling and CYPD-mediated MPTP opening.
I/R and permanent coronary occlusion induce autophagy in the myocardium through different mechanisms [160]. Myocardial I/R leads to augmentation of Beclin-1 levels, initiation of autophagosome formation, and activation of autophagy. However, reperfusion-induced autophagy plays a detrimental role, and beclin-1+/− mice display decreased infarct size. In contrast, autophagy induced by permanent coronary occlusion plays a cardioprotective role. Permanent coronary occlusion activates AMPK to inhibit a negative regulator of autophagy mTOR and thereby induces autophagy. Consistently, overexpression of dominant-negative AMPK results in attenuation of autophagy and increased infarct size [161]. However, the precise mechanisms underlying dual role of autophagy in the heart remain to be determined and require further careful investigation.
In contrast to the markedly increase in apoptosis in MI, in HF patients, cardiomyocyte apoptosis is modestly, although persistently increased [176–178]. It has been demonstrated that the Gαq-Nix/BNip3L signaling plays an important role in regulation of apoptosis during HF [179–181].
BH3-like protein BNip3 is induced during hypoxia and HF and promotes mitochondrial defects and cardiomyocyte death [182, 183]. BNip3 is an essential mediator of apoptosis and pathological remodeling following MI as evidenced by its deficiency, which leads to decreased apoptosis, remodeling, and cardiac dysfunction [184].
Growing evidence suggests that necrosis also contributes to the pathogenesis of HF. It has recently been reported that intracellular Ca2+ overload caused by transgenic overexpression of sarcolemmal L-type Ca2+ channel induces mitochondria-mediated cardiomyocyte necrosis and HF [185]. Furthermore, deletion of mediator of MPTP opening, CYPD, but not Bcl-2 overexpression, rescues this phenotype.
Elevated number of autophagosomes suggesting induced autophagy has been observed in human failing hearts [186, 187]. Analysis of atg5 −/− mice suggests that autophagy under physiological conditions is important to maintain myocardial structure and function, and induced autophagy in response to hemodynamic stress serves a protective role in HF [188]. However, study of beclin +/− mice concludes that autophagy is a pathological response to hemodynamic stress leading to HF [189].
The cardioprotective role of autophagy and lysosomal clearance is highlighted by the finding that Danon cardiomyopathy, a genetic disorder caused by defect of lysosome-associated membrane protein 2 (LAMP2), is characterized by deficiency of autophagosome–lysosome fusion [190]. Another example is patients treating with chloroquine develop a myopathic condition, which characterized by accumulation of autophagosomes [191]. Chloroquine accumulated in lysosomes neutralizes their pH and inhibits autophagosome–lysosome fusion and clearance. Thus, while autophagy normally plays cardioprotective role, “frustrated autophagy,” when autophagosome–lysosome fusion is impaired, may be detrimental [191]. Further careful investigations are required to determine whether induced autophagy plays a protective or detrimental role in myocardial disorder.
Conclusions
Living cells die in a variety of ways. A large number of stimuli can initiate apoptotic, necrotic, and autophagic cell-death pathways in the myocardium. Importantly, identical stimuli can induce different types of cardiomyocyte death. We are still in the very beginning of understanding the molecular mechanisms underlying these cellular choices. This complex, highly interconnected network contributes to the pathogenesis of various CVD including I/R injury, MI, HF, dysrhythmias, and atherosclerosis.
Due to impressive recent progress in this field, major regulators and mediators of cell death have been established. They are localized in various subcellular compartments, such as plasma membrane, mitochondria, cytosol, nucleus, ER/SR, and lysosomes, but can swiftly change their localization in response to different stimuli. Furthermore, these factors frequently play multiple roles in the intricate network of cell-death pathways contributing to its great flexibility. In addition, the extensive interconnections between different pathways of this network have made the unraveling of its nature more difficult than it had previously been anticipated.
Mitochondria are not only key regulators of myocardial cell survival providing energy but also central mediators of apoptotic and necrotic cardiomyocyte death. The key role of Bcl-2 family and BH3-only proteins in these processes has been recognized and intensively studied for the past two decades. However, identification of additional members of these families and relevant interacting partners represents an important task. The precise mechanism of interactions between Bcl-2, BH3-only, and Bak/Bax proteins is also subject of debates and intensive investigations. Moreover, these interactions are modified by membranes. Therefore, a deep understanding of how Bcl-2 family and BH3-only members act can be achieved through careful analysis of the molecular structure of these proteins as well as membranes of mitochondria and SR.
Although our understanding of the role of autophagy in the heart is rapidly evolving, its significance in myocardial disorder remains to be determined. Selective form of autophagy, mitophagy, targets damaged mitochondria to degradation and thereby can attenuate mitochondria-mediated apoptosis and necrosis in the myocardium performing a cardioprotective role. However, excessive autophagy or frustrated (uncoupled fusion with lysosomes) autophagy leads to detrimental outcomes. Recently discovered pro-apoptotic functions of a key regulator of autophagy, Beclin-1, have highlighted interconnections between different types of cell death [153]. Clearly, further investigations are required to reveal the complex role of Beclin-1 and Beclin-1-containing complexes in cardiac disease and determine if activation or inhibition of autophagy is more beneficial.
Initial attempts of therapeutic interventions in cell death have revealed that its prevention is more challenging than its induction. The most promising therapies that protect cardiomyocytes from death have been directed to the preservation of mitochondrial integrity. Small-molecule inhibitors, such as polycaspase inhibitors, UCF-101, cyclosporin A, and necrostatin-1, have shown promise in I/R, MI, and stroke in animal models and a pilot human study [4]. Trials in large animals are needed regarding applicability of these compounds to human disease. Further insight into the molecular mechanisms underlying the intricate network of cell-death pathways in the heart will increase the efficiency and repertoire of therapeutic interventions in CVD.
Summary
All multicellular organisms develop during evolution the highly regulated and interconnected processes of cell death. Cell death has initially been considered as an accidental uncontrolled process; however, it is now well recognized that many aspects of cell death are genetically programmed and therefore tightly regulated. Cell death can be classified into three types: apoptosis, necrosis, and autophagy. Mitochondria play a central role not only in the maintaining the life of cardiomyocytes by providing ATP through OXPHOS but also in regulation of cell death, responding to various stress signals.
Apoptosis is characterized by several morphological features including cell rounding-up, reduction of cellular volume (pyknosis), chromatin condensation, nuclear fragmentation (karyorrhexis), and plasma membrane blebbing. In apoptosis, plasma membrane maintains its integrity until the late stages of the process. An essential feature of apoptosis is activation of a class of specific proteases, caspases. Two distinct but interlinked pathways mediate apoptosis: the intrinsic or mitochondrial pathway and the extrinsic or death-receptor pathway.
The intrinsic pathway is induced by various stress stimuli, including growth factor deprivation, oxidative and genotoxic stress, hypoxia, and various toxins. These signals are transduced by the Bcl-2 family proteins to mitochondria, leading to release of mitochondrial apoptotic proteins, and to sarcoplasmic reticulum (SR), resulting in release of Ca2+.
The Bcl-2 proteins are characterized by the presence of the Bcl-2 homology (BH) domains. The anti-apoptotic Bcl-2, Bcl-XL, Bcl-w, Mcl-1, A1, and Boo contain four BH domains, BH1–BH4. The pro-apoptotic Bax and Bak proteins also contain four BH domains. The BH3-only proteins (Bim, Bid, Puma, Bad, Noxa, Bmf, Bik, and Hrk), containing only the BH3 domain, are able to bind and inhibit the pro-survival Bcl-2 proteins, releasing thereby the pro-apoptotic Bax/Bak.
In response to apoptotic signals, Bax and Bak undergo conformational activation leading to their recruitment to the MOM. At the mitochondria, these multi-domain proteins form homo- and hetero-oligomers, resulting in generation of the “apoptotic pore” in the MOM, which represents the point of no return in mitochondrial apoptotic pathway.
Permeabilization of the MOM promotes the release of multiple apoptogens (e.g., cytochrome c, Smac/DIBLO, endoG) from the mitochondrial intermembrane space into the cytosol. Released cytochrome c along with the cytosolic dATP binds to Apaf1 inducing its oligomerization and formation of a heptameric protein complex, apoptosome. The apoptosome can bind procaspase 9 promoting its cleavage and activation. Activated caspase 9 in turn cleaves and activates numerous downstream procaspases, proteases, and DNases resulting eventually in the cell demolition.
Several proteins belonging to the IAP family can bind and inhibit caspases. In addition, IAPs due to their E3 ubiquitin ligase activity can promote proteasomal destruction of caspases in response to apoptotic stimuli. Other released mitochondrial pro-apoptotic factors, Smac/DIABLO and Omi/HtrA2, can bind IAPs releasing thereby the caspases.
Extrinsic apoptotic pathway also known as the death-receptor pathway is activated by the binding of death ligands, such as FasL, TNF, or TRAIL, to their cognate cell surface death domain-containing receptors, known as death receptors. They share common activation mechanism: binding of specific ligands to their respective receptors induces receptor homo-trimerization, which triggers the recruitment of cytosolic adaptor and effector proteins. This in turn recruits procaspase 8 or 10 to assemble the multiprotein DISC. DISC formation induces activation of caspase 8 or 10, which in turn cleave and activate downstream effector procaspases, primarily procaspases 3, 6, and 7.
Although the DISC-mediated direct activation of effector procaspases may be sufficient in some cells (type I cells), in most cells (type II cells) activation of the BH3-only protein Bid is required for the efficient apoptotic response. The DISC-activated caspase 8 cleaves Bid into a pro-apoptotic, active, truncated form, tBid. tBid translocates to mitochondria and induces MOM permeabilization leading to release apoptogens and to the mitochondria-dependent apoptosis. Thus, tBid not only facilitates the extrinsic pathway but also provides the link between the death-receptor and mitochondrial apoptotic pathways.
Necrosis is characterized morphologically by cell and organelle swelling (oncosis), early plasma membrane rupture, and resultant loss of intracellular contents. While this type of cell death has been viewed, in contrast to apoptosis, as a purely accidental uncontrolled process, emerging evidence suggests that many aspects of necrosis could be finely regulated. Over the past decade, it has been demonstrated that activation of death receptors can initiate not only extrinsic apoptotic pathway but also necrosis.
The binding of TNF-α to TNF-R1 triggers the assembly of the membrane-proximal complex I that includes also TRADD, RIP1, cIAP1 and 2, and TRAF2 and 5. cIAPs are E3 ubiquitin ligases, which catalyze Lys63 polyubiquitination of RIP1 and TRAF2. Polyubiquitinated RIP1 and TRAF2 bind TAK1 along with TAB2 and 3. The formed TAK1–TAB2/3 complex mediates the canonical NF-κB activation pathway, which transactivates multiple pro-survival genes. However, complex I can mediate also the recruitment of the ROS-generating NOX1 to the plasma membrane contributing thereby to necrosis.
Internalization of complex I results in dissociation of TNF-R1, deubiquitination of RIP1 by CYLD, and recruitment of FADD and procaspase 8. Resultant multiprotein signaling complex is known as complex II. Within complex II, procaspase 8 is activated and can promote apoptosis by activating the caspase cascade. Caspase 8 can also cleave and inactivate RIP1 and 3 inhibiting thereby either necrosis or pro-survival NF-κB-mediated pathway. However, if caspase 8 is inhibited or deleted, RIP1 can recruit RIP3 to the complex, where they are phosphorylated, and complex II promotes necrosis.
The opening of the MPTP is another essential mechanism underlying necrosis. MPTP opening is induced by excessive OS and elevated Ca2+ and phosphate concentrations and adenine nucleotide depletion. This leads to the dissipation of the Δψm, inhibition of ATP synthesis, increased ROS generation, and eventually in swelling and rupture of the mitochondrion. CYPD is a critical component of the MPTP, which regulates its opening. PARP, Ca2+, and non-caspase proteases, calpains and cathepsins, represent other important mediators of necrotic cell death.
Autophagy is morphologically characterized by the massive formation of single- or double-membrane lysosomal-derived vesicles, which contain degenerating cytoplasmic particles, organelles, and protein aggregates. Autophagy is primarily a survival response to nutrient deprivation to recycle intracellular components and provide the cell with metabolites. It is also a protective mechanism to eliminate damaged cellular organelles and toxic misfolded proteins.
mTOR and Beclin-1 are two main regulators of autophagy. mTOR is the central sensor of nutrient status, which is activated upon fed conditions to stimulate protein synthesis and inhibit autophagy via phosphorylation and inactivation of Atg. AMPK can inhibit mTOR and thereby override mTOR-mediated suppression of autophagy. In response to starvation, class I PI3K signaling promotes mTOR inhibition resulting in suppression of protein synthesis and activation of autophagy.
The p53 tumor suppressor can control autophagy in dual fashion: basal p53 levels inhibit AMPK relieving mTOR-mediated suppression of autophagy, while p53 activation induces autophagy through AMPK activation and transcriptional upregulation of PTEN, DRAM, and TSC1. Importantly, activation of p53 by distinct stress stimuli induces apoptosis through engagement of Puma and Noxa.
Human BH3-only protein Beclin-1 in association with PI3K3C/Vps34, Vps15, and possibly Ambra1 forms the core complex. Binding of Atg14 and possibly UVRAG and Bif-1 activates the core complex to initiate vesicle nucleation. While Atg14, UVRAG, and Bif-1 activate autophagosome formation, recently identified component of Beclin-1–Vps34–Vps15 complexes, Rubicon, negatively regulates this process.
A complex cascade of the Atg-ubiquitin conjugation reactions mediates elongation of the pre-autophagosome and attachment to damaged organelles and protein aggregates. Maturation of the pre-autophagosome followed by its closure generates the double-membrane autophagosome. The mature autophagosome fuses with lysosome delivering its contents for destruction by the lysosomal enzymes.
While autophagy has initially been viewed as a nonselective process for degradation damaged organelles and protein aggregates, it becomes clear that molecular mechanisms exist for the selective recruitment of the cargo to autophagosomes. The term “mitophagy” has been introduced to describe selective autophagic removal of damaged mitochondria. Recent studies suggest a number of factors such as Atg32, Parkin, PINK1, BNip3, and Nix (also called BNip3L), which confer selectivity during mitophagy. However, the mechanisms by which these factors mediate this process remain to be determined.
Both components of the autophagy-promoting core complex, Beclin-1 and PI3K3C/Vps34, can be cleaved by caspases 3, 7, and 8 in response to activation of both mitochondrial and death-receptor apoptotic pathways. In contrast to full-length Beclin-1, its N- and C-terminal cleavage fragments fail to induce autophagy. More importantly, the C-terminal fragment of Beclin-1 acquires a new function facilitating mitochondria-mediated apoptosis. Thus, Bcl-2/Bcl-XL–Beclin-1 interactions represent not only a key mechanism to switch on or off autophagy but also an important link between different cell-death pathways.< div class='tao-gold-member'>Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


