Cryptococcosis and the Endemic Mycoses
Histoplasmosis and blastomycosis mostly afflict those living in the Mississippi and Ohio River Valleys, while coccidioidomycosis occurs primarily in the Southwestern desert of the United States (Fig. 134-1). Histoplasmosis and coccidioidomycosis also are endemic in parts of Mexico and Central and South America. Infection with Cryptococcus neoformans var. gatti is endemic to parts of British Columbia, the Pacific Northwest region of the United States and Australia while C. neoformans var. neoformans exhibits no geographical predilection. These mycoses are often mistakenly diagnosed and incorrectly treated as community-acquired pneumonia or sarcoidosis, resulting in serious morbidity or death in many cases.1–3
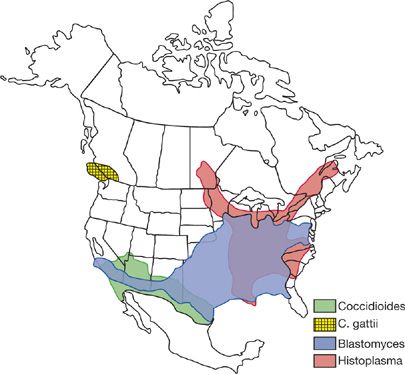
Figure 134-1 Geographical distribution of endemic mycoses in the United States. The area of histoplasmin skin test reactivity in the southwestern US represents cross-reactivity caused by coccidioidomycosis, not endemic histoplasmosis.
These organisms are found in the soil, and infection occurs following inhalation of the infectious forms of these fungi when sites containing the organism are disturbed. In some endemic areas over one-half of residents have acquired these mycoses early in life. In healthy individuals the infections are usually asymptomatic or clinically self-limited. In others, especially those who are immunosuppressed, the course of infection may be progressive and fatal without treatment. While these mycoses are less often seen in patients with acquired immunodeficiency syndrome (AIDS) who have access to effective antiretroviral therapy, they are occurring more frequently in those with other immunosuppressive conditions, and continue to be pathogenic for individuals without underlying disease. The majority of those hospitalized due to infection with endemic mycoses in the United States in fact have no identifiable immunologic deficits. In a study of patients hospitalized with histoplasmosis, blastomycosis, or cocciodiodomycosis, only 13% of those infected were immunosuppressed. Hospital costs alone exceeded $250 million,4 providing an estimate of the impact of the endemic mycoses in the United States. Advances in diagnosis and treatment provide opportunities to improve the outcome of these mycoses.
CRYPTOCOCCAL INFECTIONS
Cryptococcosis is caused by infection with the encapsulated fungus C. neoformans, an organism with a worldwide distribution. Inhalation of C. neoformans initiates the infection in the lung with hematogenous dissemination most often involving the meninges. Although pulmonary infection may be discovered in the presence or absence of disseminated infection, meningoencephalitis remains the most commonly diagnosed form of cryptococcal infection. The spectrum of disease ranges from asymptomatic pulmonary infection in the immunocompetent host to diffuse pulmonary disease associated with respiratory failure and widespread disseminated disease in the immunocompromised host. It is estimated that nearly 1 million cases of cryptococcal meningitis occur worldwide annually, with the majority of cases occurring in those with HIV infection.5 The incidence of cryptococcosis in patients with AIDS in the United States has declined since the introduction of potent antiretroviral therapy; most cases now occur in those with limited access to care.5,6
 MYCOLOGY
MYCOLOGY
C. neoformans is a yeast that is characterized by a thick polysaccharide capsule. The yeast measures 4 to 6 μm in diameter but the capsule thickness varies from 1 to more than 30 μm. Organisms are smaller and less well encapsulated in the environment, explaining the ability to reach the terminal airways following inhalation. C. neoformans grows readily in fungal media, allowing isolation in less than 48 hours and identification by biochemical tests or DNA probes. Four serotypes of C. neoformans have been described—A, B, C, and D.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Serotypes A and D predominate in North America and Europe and grow best in composted bird droppings or rotted vegetation. Serotypes B and C are classified as C. neoformans var. gatti and are more common in tropical and subtropical regions in association with eucalyptus trees rather than avian excreta. Recently outbreaks of infection caused by C. neoformans var. gatti have been reported in British Columbia and the Pacific Northwest region of the United States (Fig. 134-1). Otherwise outbreaks or clusters of cases are rare in cryptococcosis, and in most cases a history of exposure to birds or dust is lacking. Person-to-person transmission does not occur if cryptococcosis has been transmitted through organ transplantation; cutaneous infection has occurred after direct inoculation.
Patient populations at increased risk for progressive cryptococcosis include those with T-cell–mediated immune defects caused by AIDS, lymphoreticular malignancy (particularly Hodgkin disease), or immunosuppressive medications, including tumor necrosis factor (TNF) inhibitors. The disease also appears to be more frequent in diabetics. While C. neoformans var. gatti infection was initially reported as occurring almost exclusively in immunocompetent hosts, in a recent surveillance study, half of patients infected with this organism had underlying immunosuppression.7
 PATHOGENESIS
PATHOGENESIS
Cryptococcosis is acquired by inhaling aerosols containing the yeast but rarely by direct inoculation. Progressive disease often follows exposure in patients with impaired cellular immunity. In tissues, a mixed macrophage, lymphocyte, and plasma-cell response is seen, but inflammation may be minimal in immunodeficient subjects. Granulomas are uncommonly found in the nervous system but may be seen in other tissues.
Macrophages, natural killer cells, and T lymphocytes play the key roles in cellular defense against C. neoformans. Inflammatory cytokines (interleukin [IL]-2, IL-12, interferon [IFN]-γ) and macrophage colony-stimulating factor enhance the antifungal activity of these cellular mechanisms. Humoral immunity complements cellular mechanisms in defense against C. neoformans.
 CLINICAL FINDINGS
CLINICAL FINDINGS
Cryptococcal infection results in asymptomatic or self-limited pulmonary disease in most healthy individuals. While symptomatic isolated pulmonary cryptococcosis may be diagnosed, meningoencephalitis is the most commonly recognized manifestation of cryptococcosis and the most common cause of death. Hematogenous dissemination to almost any tissue occurs in fewer than 25% of cases.
Pulmonary
Isolated pulmonary infection is not uncommon, and saprophytic colonization has been observed. Pulmonary cryptococcosis often is asymptomatic, discovered when chest radiographs are done for other reasons. Concurrent disseminated disease occurs in about 15% of cases. In symptomatic cases, common complaints include dry cough, dull chest discomfort, and low-grade fever. Less commonly night sweats, fatigue, weight loss, or hemoptysis may occur. Nodular infiltrates are typical of pulmonary cryptococcosis in the normal host (Fig. 134-2). Pulmonary cryptococcosis in nonimmunosuppressed patients usually resolve without therapy. Occasionally pulmonary cryptococcosis progresses slowly or may be accompanied by meningoencephalitis or dissemination to other organs after improvement of the pulmonary process.
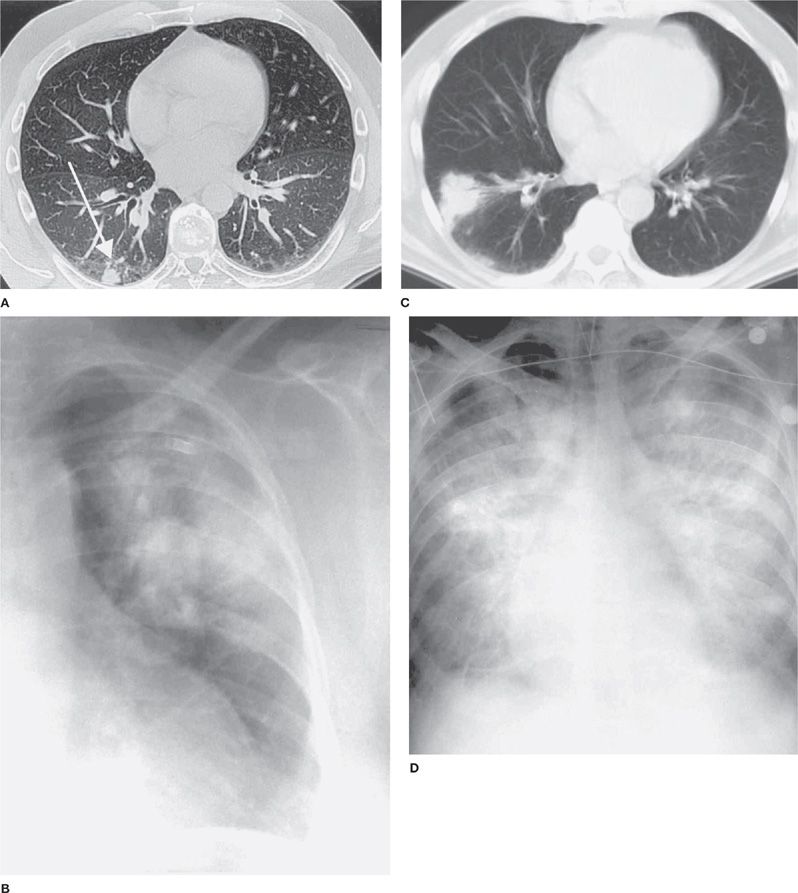
Figure 134-2 Chest radiograph findings of cryptococcosis. A. Peripheral nodule. B. Mass-like lesion. C. Computed tomography of mass lesion. D. Chest radiograph showing diffuse infiltrate.
In contrast to nonimmunocompromised patients, most immunocompromised patients exhibit fever and cough. Diffuse interstitial infiltrates or widespread alveolar consolidation causing respiratory failure is common in severely immunodeficient hosts. Radiographs in patients with less profound cell–mediated immune defects usually show nodular or patchy alveolar infiltrates (Fig. 134-2). Mortality is high in immunosuppressed patients with diffuse pulmonary involvement. Mass lesions are not uncommon and may resemble malignancy. Cavitation is uncommon and mediastinal adenopathy, pleural effusion, and calcification are rare. A halo sign, usually attributed to aspergillosis, may be observed in cryptococcosis. Empyema, pleural disease suggesting a Pancoast tumor, and pneumothorax have been reported. As immunosuppressed patients with pulmonary cryptococcosis often have meningoencephalitis, a lumbar puncture is recommended in these patients even in the absence of signs or symptoms of central nervous system (CNS) infection.
Meningoencephalitis
Meningoencephalitis is the most common manifestation of cryptococcosis. A gradual onset is typical, but a more rapid presentation may be seen in patients with severe immunodeficiency. Symptoms include fever, headache, nausea, and vomiting, while less than one-third of patients exhibit meningismus, altered mentation, or focal neurological abnormalities. Elevated intracranial pressure is common and may cause brain-stem herniation. Focal brain lesions occur in about 10% of cases, alone or in combination with meningoencephalitis.
Other Sites of Dissemination
Extraneural involvement may be seen in patients with meningoencephalitis or pulmonary cryptococcosis. Hepatosplenomegaly and bone marrow suppression are seen most commonly while lesions involving the skin, eyes, bones, or joints occur in about 5%. Other sites of dissemination include the heart, pericardium, muscle, gastrointestinal tract, peritoneum, thyroid, larynx, breast, placenta, urinary tract, prostate, and organ of Corti.
Immune Reconstitution Syndrome
Up to 30% of patients with AIDS and a recently diagnosed cryptococcal infection may exhibit an immune reconstitution inflammatory syndrome (IRIS) following initiation of highly active antiretroviral therapy (HAART) (see Chapter 123).8 In AIDS patients, levels of serum cryptococcal antigen titer9 as well as CSF cryptococcal antigen titers8 prior to the administration of HAART have been positively correlated with the risk of IRIS. Similar findings have been reported following organ transplantation with reductions in immunosuppression during therapy. Clinical findings have included worsening of meningitis with the development of intracranial hypertension, hypercalcemia, intrathoracic lymphadenopathy, cavitation of pulmonary lesions, and soft tissue abscess. These findings represent inflammation resulting from an enhanced inflammatory response made possible by immune reconstitution. While such changes may resolve spontaneously at times, the inflammatory changes within the CNS have led to death.10 A recently published study that randomized patients with newly diagnosed AIDS and cryptococcal meningitis to early HAART (approximately 7 days following initiation of antifungals) versus delayed HAART approximately (28 days after initiating antifungals) demonstrated significantly more IRIS events in those treated with early HAART.11 These findings would support the recommendation by the Infectious Disease Society of America (IDSA) that HAART should be withheld for individuals recently diagnosed with cryptococcal meningitis, for 2 to 10 weeks after initiation of antifungal therapy.12
 DIAGNOSIS
DIAGNOSIS
Approaches to the diagnosis of cryptococcal pneumonia and meningoencephalitis are discussed below.
Pneumonia
The diagnosis of pneumonia is usually made by cytology or histopathology of respiratory secretions or lung tissue, and confirmed by culture (Table 134-1).13–15 Once a diagnosis of pulmonary cryptococcosis is made, an evaluation for extrapulmonary dissemination should be initiated. A serum cryptococcal antigen test should be performed along with fungal blood culture. Cerebrospinal fluid (CSF) examination is recommended if the patient has any symptoms of meningitis or brain involvement, or is immunosuppressed. IDSA guidelines indicate that for immunocompromised patients with pulmonary cryptococcosis, a lumbar puncture to rule out asymptomatic CNS involvement should be considered.12 However, for normal hosts with an asymptomatic pulmonary nodule or infiltrate, no CNS symptoms, and a negative or very low serum cryptococcal antigen, a lumbar puncture can be avoided.
TABLE 134-1 Diagnostic Studies in Cryptococcosis (Percent Positive Assays)
Histopathology and Cytology C. neoformans can be recognized in tissue as a globose or oval to lemon-shaped yeast with a polysaccharide capsule (Fig. 134-3). Cryptococci are readily identified by the Gomori methenamine silver (GMS) and periodic acid–Schiff (PAS) stains. More specific stains for C. neoformans include the Mayer’s mucicarmine stain, which stains the fungal capsule and the Masson–Fontana melanin stain, which may detect capsule-deficient cryptococci. Direct microscopic examination of bronchoalveolar lavage (BAL) fluid sediment stained with India ink can identify the organism. The sensitivity of histopathology in patients with AIDS is about 67%. In the nonimmunocompromised host, fungal stains of respiratory specimens are rarely positive (approximately 5%) despite isolation of the fungus in over two-thirds of cases.
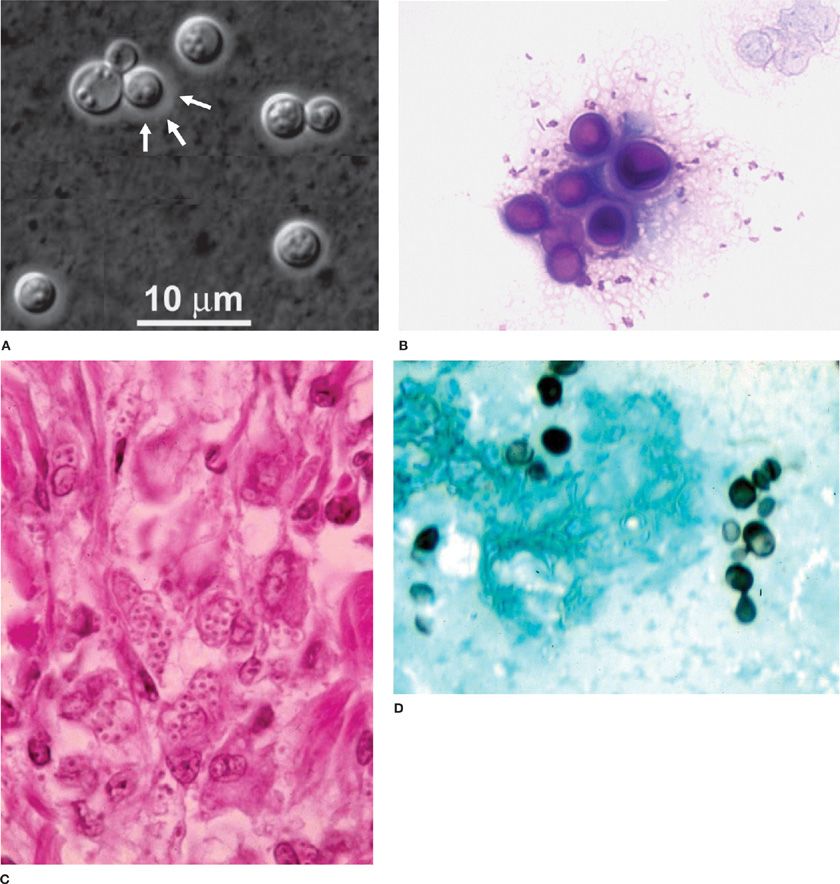
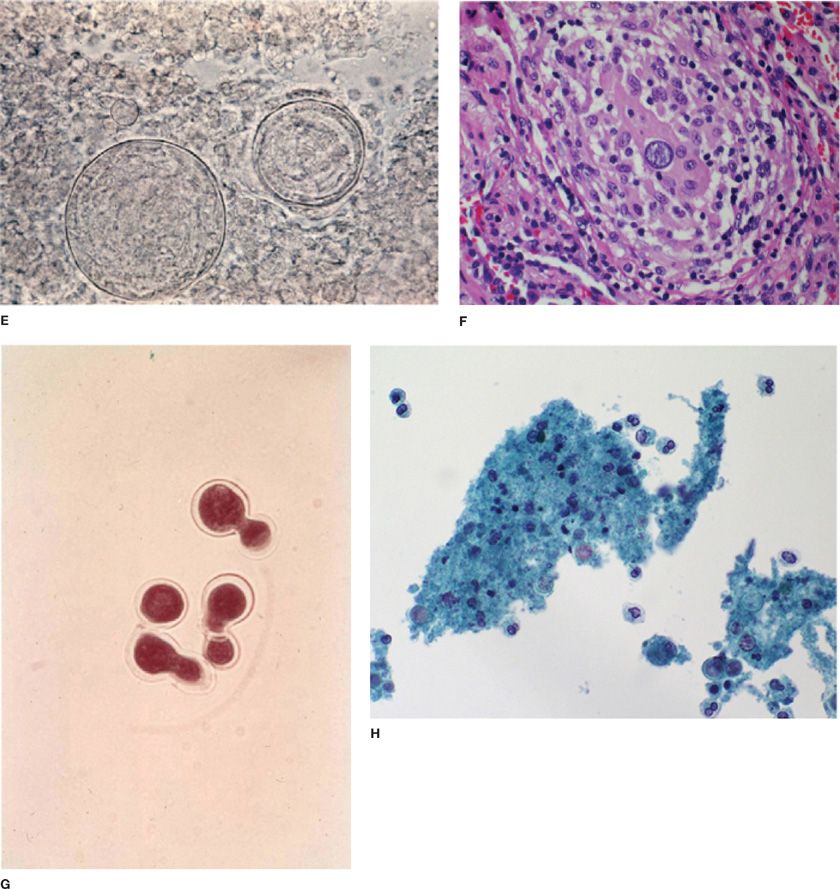
Figure 134-3 Fungal histopathology. A. India ink wet mount C. neoformans. B. Periodic acid–Schiff (PAS) stain of C. neoformans. C. Hematoxylin and eosin (H&E) stain H. capsulatum. D. Gomori methenamine silver stain of H. capsulatum yeast. E. Potassium hydroxide (KOH) wet mount showing C. immitis spherule. F. H&E stain C. immitis spherule in the tissue. G. KOH wet mount showing B. dermatitidis yeast. H. Papanicolaou stain showing B. dermatitidis yeast.
Antigen Detection In patients with cryptococcal pneumonia, the serum cryptococcal antigen is usually not detected unless extrapulmonary dissemination has occurred, the pneumonia is extensive, or the patient is immunosuppressed. A negative serum cryptococcal test should not be used to exclude the diagnosis of cryptococcal pneumonia. In patients with AIDS with pulmonary cryptococcosis the serum cryptococcal antigen is positive in most cases. Cryptococcal antigen also may be detected in BAL and pleural fluid. Cryptococcal antigen detection using a lateral flow device simplifies point-of-care testing.19 Cryptococcal serum antigen screening of patients with HIV-infection and a CD4 count <100 cells/mm3, who reside in an area with a high rate of cryptococcal disease, can identify those at risk to develop cryptococcal meningitis over the subsequent year.20
Culture C. neoformans often can be isolated from sputum in patients with cryptococcal pneumonia. However, isolation from sputum may represent mere colonization. In patients unable to produce sputum, bronchoscopy may be useful. Cultures of BAL or lung tissue yield the organism in 50% to 90% of cases while fungal stains are positive less often. Needle aspiration of focal lesions also may be useful. Thoracentesis may yield the organism in patients with pleural involvement. Blood cultures may be positive in immunocompromised hosts.
Meningoencephalitis
The diagnosis of meningitis can be made initially by India ink stain or detection of antigen in CSF and confirmed by isolation of the organism from fungal cultures. Antigen also can be detected in serum, providing a clue to the diagnosis before lumbar puncture is performed. Antigen may be detected in the serum in 92% to 100% and the urine in 98% to 100% of immunocompromised patients with cryptococcal meningitis,16–18 but in a lower proportion of nonimmunocompromised patients.21 Cultures also may be positive from extrapulmonary sites in up to two-thirds of patients.
 TREATMENT
TREATMENT
Treatment is indicated in patients with symptomatic pulmonary infections, especially if they are immunocompromised and in all patients with meningoencephalitis or disseminated infection (Table 134-2). The rationale and supporting evidence for these recommendations are discussed in an IDSA guideline document published in 2010.12 Patients with a diagnosis of cryptococcosis at any site should be provided close follow-up for at least 1 year as the majority of relapses occur during this time period.
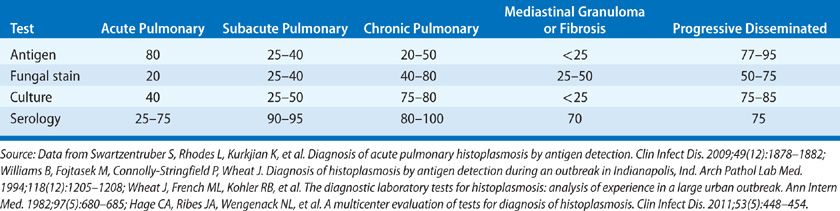
TABLE 134-2 Diagnostic Studies in Histoplasmosis (Percent Positive Assays)
Indications for Treatment
For asymptomatic and immunologically normal patients without extrapulmonary disease (e.g., pulmonary nodules), antifungal therapy may be withheld for up to a month if patients can be followed closely. Increasing size or numbers of lesions would be indications for the treatment. For symptomatic patients, those who are immunocompromised, and patients showing progression during observation, treatment is advised.
Treatment of meningoencephalitis and and extrapulmonary dissemination (even without meningoencephalitis) is indicated in all cases.
Selection of Antifungal Agent
Selection of an antifungal agent for treatment is considered below for pneumonia, meningoencephalitis, and disseminated disease.
Pneumonia For nonimmunosuppressed patients, fluconazole 400 mg/d for 6 to 12 months is recommended, and itraconazole (200 mg twice per day orally), voriconazole (200 mg twice per day orally), and posaconazole (400 mg twice per day orally) may be considered acceptable alternatives only if fluconazole is unavailable or contraindicated.12 In more severe cases, amphotericin B and flucytosine should be given as recommended for meningitis (see below). Treatment guidelines for meningitis should be followed in immunosuppressed patients.
Meningitis Amphotericin B 0.7 to 1.0 mg/kg/d and 5-flucytosine 100 mg/kg/d are favored and should be given for 2 weeks (induction) followed by fluconazole 400 mg/d for 10 weeks (consolidation). 5-flucytosine accelerates sterilization of the CSF, improves the overall response rates for meningitis and reduces the risk for relapse. Lipid preparations of amphotericin B (3–5 mg/kg/day) are less toxic and may be preferred over the standard formulation, notably in patients with renal impairment or those who are thought to be at increased risk for amphotericin B nephrotoxicity. Amphotericin-containing preparations with or without flucytosine for 4 to 6 weeks without additional therapy can be potentially curative in patients with negative cultures after 2 weeks of therapy who are without any immunosuppression, neurologic complications or meningeal symptoms, though for most some form of antifungal consolidation therapy is recommended. Fluconazole alone is not recommended as induction therapy in immunocompromised hosts when amphotericin and 5-flucytosine are available. Combinations of therapy using amphotericin B and fluconazole at various doses as induction therapy have been studied in cryptococcal meningitis in resource poor settings where 5-flucytosine cannot be administered.26,27 Compared to amphotericin B alone, combined amphotericin B plus fluconazole was associated with a trend toward improved outcomes.12 Improved responses using this combination have been associated with a daily dose of fluconazole of 800 mg rather than 400 mg12,27 and correlated with the ability to achieve higher CSF and serum fluconazole levels.27 Fluconazole 800 to 1200 mg daily given with 5-flucytosine has been studied as induction therapy and when compared to treatment with fluconazole alone this combination appears to be associated with improved responses though greater toxicities.28
Fluconazole alone as induction therapy can be considered when amphotericin and 5-flucytosine are not available as is not uncommon in resource-limited settings. Induction doses of fluconazole of 1200 mg daily are associated with higher response rates than 800 mg fluconazole daily in a recent study.29 Itraconazole 200 mg twice daily is an alternative in patients unable to take fluconazole for consolidation therapy.
Aggressive management of elevated intracranial pressure through removal of large volumes (about 25 mL) of CSF is essential to achieve the highest survival. Addition of IFN-γ led to clinical improvement in two patients with CD4 lymphopenia unresponsive to amphotericin B.
In patients with active AIDS or other immunosuppressive conditions, so-called maintenance or suppressive treatment with fluconazole 200 mg/d is recommended. Itraconazole is inferior to fluconazole for chronic maintenance therapy and is not advised. If itraconazole is used, 200 mg twice daily is recommended. Amphotericin B at a dose of 1 mg/kg one to three times weekly is another alternative maintenance regimen.
With the use of HAART, studies have indicated that maintenance fluconazole can be safely discontinued in most patients responding to HAART with a CD4 cell count >100 cells/mm3, an undetectable or low HIV RNA level sustained for 3 months, and at least 1 year of antifungal drug treatment. Patients require careful monitoring for relapse and continued response to HAART. Resumption of antifungal therapy is recommended if the CD4 count falls below 100 cells/mm3. Monitoring serum cryptococcal antigen should also be performed, as a rising titer would support resumption of antifungal therapy.12
For immunosuppressed patients without AIDS, a 6- to 12-month course of fluconazole is recommended. As in patients with AIDS, careful follow-up is required after antifungal therapy is stopped. In some patients who relapse following such a course of therapy, lifelong maintenance therapy may be appropriate.
Disseminated/Extrapulmonary, without Meningoencephalitis Trials have not been performed evaluating treatment of non-CNS extrapulmonary disease, and recommendations are based upon experience of the IDSA guideline committee. Recommendations outlined earlier for pneumonia should be followed in nonimmunosuppressed patients and for meningitis in those who are immunosuppressed.
Role of Antifungal Resistance
Resistance may be a cause for fluconazole failure. The minimum inhibitory concentration (MIC) of fluconazole required to inhibit greater than 90% of strains of C. neoformans is likely somewhere between 8 and 16 μg/mL, which can be used as a breakpoint for defining resistance.30 In one study from Spain, patients infected with C. neoformans isolates with MICs ≥16 μg/mL failed to respond to fluconazole therapy.31 In a recent study from Africa evaluating relapses of meningitis in individuals treated with primary fluconazole therapy alone, 76% of relapses associated with positive cultures demonstrated resistance to fluconazole with MICs to fluconazole of ≥64 μg/mL.32 It is recommended that testing of isolates for fluconazole resistance should be reserved for those who do not respond to therapy, those who relapse and those who develop cryptococcal meningitis after prolonged exposure to azole therapy.12 As 5-flucytosine susceptibilities have not been demonstrated to be predictive of response, such testing is not routinely recommended. While there are no recommendations to routinely tests isolates for primary amphotericin B resistance, reduced susceptibility of C. neoformans isolates to amphotericin B was associated with greater day 14 mortality in one study of meningitis treatment in AIDS patients treated with amphotericin B alone as induction therapy.33
Newer Antifungal Agents
Alternatives to itraconazole and fluconazole are needed for patients who fail or do not tolerate those agents. Voriconazole is slightly more active in vitro than posaconazole, with an MIC90 levels in the range of 0.12 to 0.25 μg/mL.34 Voriconazole penetrates CSF, but not as well as fluconazole. Furthermore, strains with high-level resistance to fluconazole may be cross-resistant to voriconazole.35 Posaconazole was effective in a rabbit model of cryptococcal meningitis.36 While not studied adequately case reports from salvage studies support the potential effectiveness of voriconazole37 as well as posaconazole.38 Neither would appear to offer advantages over fluconazole, however as primary therapy. The echinocandins are not active in vitro,39 have not been studied in animal models, and are not recommended in patients with cryptococcal infection.
Adjunctive Therapy
Adjunctive IFN-γ immunotherapy has been used in randomized controlled trial in patients with cryptococcal meningitis receiving amphotericin B and 5-flucytosine.40 While no mortality benefit was seen in those who received IFN-γ in addition to standard antifungal treatment, clearance of C. neoformans from the CSF was noted to be significantly faster in those who were treated with adjunctive IFN-γ.40 Given the expense of this adjunctive therapy and lack of mortality benefit, routine use of IFN-γ for cryptococcal meningitis would appear to be unwarranted. Some however have recommended consideration of this adjunctive therapy or refractory cases.12
 PREVENTION AND SCREENING FOR CRYPTOCOCCOSIS
PREVENTION AND SCREENING FOR CRYPTOCOCCOSIS
Although fluconazole and itraconazole reduced the incidence of cryptococcosis in patients with AIDS, prophylaxis is not recommended in resource rich settings, largely because of the low attack rate. In such regions, unlikely cost-effectiveness, limited impact on survival, and potential to induce fluconazole resistance among other fungi are reasons this strategy is not recommended. On the other hand in areas where cryptococcal infections are much more common and associated with high mortality prophylaxis may reduce mortality and represent an effective strategy.41 Serum cryptococcal antigen screening on all HIV-infected individuals with CD4 counts less than 100 cells/mm3 living in resource-limited settings characterized by high rates of cryptocccal meningitis, can identify those at high risk for clinical cryptococcal disease.42 It has been suggested that identifying such individuals who may benefit from antifungal therapy in the absence of clinical disease may represent a cost-effective strategy.41,42
HISTOPLASMOSIS
Histoplasmosis is the most common endemic mycosis and a major cause of morbidity in patients who live in endemic areas. Progressive disseminated histoplasmosis (PDH) is an important complication of AIDS, immunosuppression for the prevention of transplant rejection,43 treatment of inflammatory diseases44,45 or malignancies, and immunodeficiency disorders.46 Understanding of the clinical syndromes and untreated course of the infections is essential in the diagnosis and management of patients with histoplasmosis. Improved laboratory tests have made it possible to rapidly diagnose the more severe cases. Expanded treatment options are available using newer triazole antifungal agents and liposomal formulations of amphotericin B.
 MYCOLOGY
MYCOLOGY
Histoplasma capsulatum grows as a mold in the soil and converts to a yeast in tissues (Fig. 134-3). Microconidia measuring 2 to 4 μm in diameter are the infectious particle in the mold phase of the organism. H. capsulatum also grows as a mold on fungal media in the laboratory. Definitive identification of a mold as H. capsulatum is made by DNA probe or exoantigen detection. At temperatures above 35°C, H. capsulatum grows as a yeast, which is the pathogenic form found in the tissues. The yeast measures about 2 to 5 μm and exhibits narrow-based budding.
 EPIDEMIOLOGY
EPIDEMIOLOGY
H. capsulatum is endemic in areas of North and South America (see Fig. 134-1) but can be found throughout the world. Bird and bat excrement enhance growth of the organism by accelerating sporulation. Cases outside the endemic region may have been acquired by exposure during travel or prior residence in the endemic area, or exposure to microfoci containing the organism within the nonendemic area. In the endemic areas between 10% and 90% of individuals exhibit histoplasmin skin test positivity as evidence of past histoplasmosis.47
 PATHOGENESIS
PATHOGENESIS
Infection develops when conidia are inhaled and germinate into yeast. In many cases primary infection is asymptomatic and goes undiagnosed. Clinical illness most often follows exogenous infection or reinfection. With the development of cell-mediated immunity during the first month following initial infection, IFN-γ and IL-12 arm macrophages to kill the fungus and halt progression of the disease.48 TNF-α also is very important for immune defense against H. capsulatum.49 These defense mechanisms are sufficient to control the infection in immunocompetent individuals, explaining the subclinical or self-limited course characteristic of acute histoplasmosis.
Reactivation of latent infection may account for some cases occurring in individuals with past histoplasmosis who become immunocompromised. Studies showing a low incidence of histoplasmosis following immunosuppression in children,50 organ transplantation,51 or treatment with TNF inhibitors,44 however, suggest reactivation is rare. Also, latency could not be demonstrated experimentally in calcified lung lesions identified as incidental findings at autopsy.52
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
Heavy exposure can cause severe illness even in healthy subjects. Low-level exposure is more common, however, and usually causes asymptomatic infection or clinically self-limited infection. The common self-limited presentations include acute and subacute pulmonary histoplasmosis, pericarditis, and rheumatological syndromes.
Asymptomatic Infection
In endemic areas, between 10% and 90% of residents exhibit histoplasmin skin test reactivity by age 20.47 In most of these cases, infection was presumed to have been asymptomatic. Most asymptomatic cases are identified as an incidental finding on chest radiographs or computed tomography (CT) scans performed for other reasons. The most common findings are enlarged mediastinal or hilar lymph nodes or pulmonary nodules.1 The main significance of such findings is the need to differentiate them from malignancy or tuberculosis. Asymptomatic infection also may be identified by demonstration of seropositivity during evaluation of another condition.
Pulmonary Syndromes
A variety of pulmonary syndromes may be seen with histoplasmosis. Each is considered below.
Acute Pulmonary Histoplasmosis
Following heavy exposure, patients present with diffuse pulmonary involvement often causing respiratory insufficiency.1,53 Chest radiographs usually show diffuse interstitial, reticulonodular infiltrates, nodular, or patchy airspace disease, but a miliary pattern suggestive of hematogenous dissemination may be seen (Fig. 134-4). Initial radiographs may appear normal, evolving to show infiltrates over the next few weeks. Mediastinal adenopathy is common. Evidence for extrapulmonary dissemination is present in about 40% of cases.22 While patients may recover without therapy,54 recovery may be slow,55 respiratory failure may develop,56 and some infections may be fatal.56,57
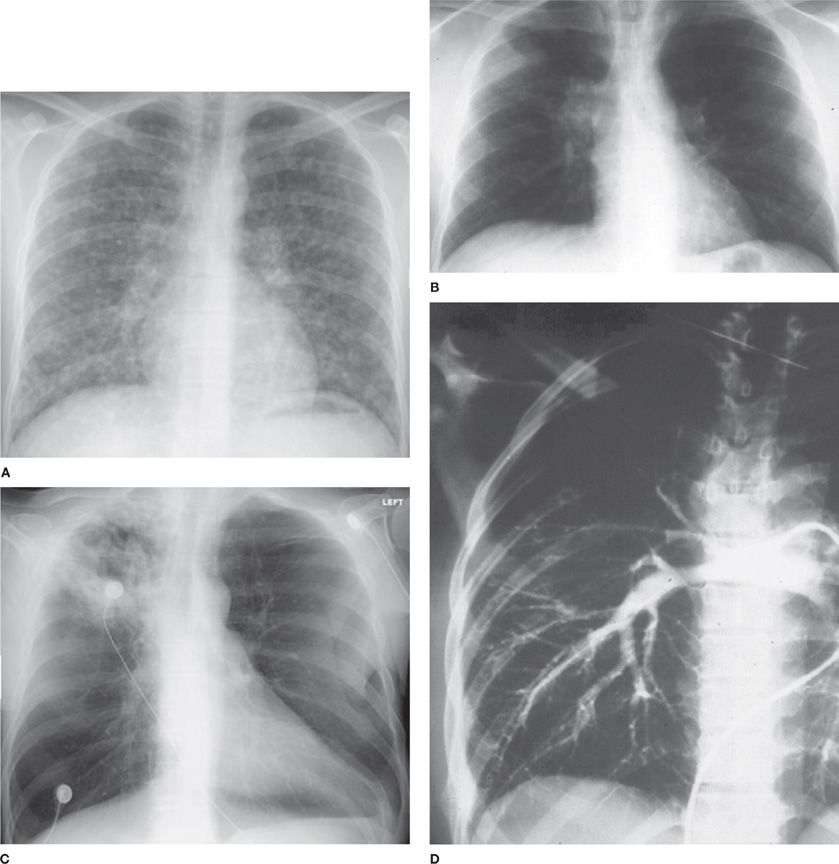
Figure 134-4 A. Chest radiograph of acute diffuse histoplasmosis after large inoculum exposure. B. Computed tomography of subacute histoplasmosis with mediastinal adenopathy. C. Chest radiograph of cavitary histoplasmosis. D. Pulmonary arteriogram showing obstruction of pulmonary artery to right upper lung caused by fibrosing mediastinitis.
Subacute Pulmonary Histoplasmosis More commonly, the inoculum is small and the presentation is subacute.1,53 Patients present with fever, cough, and chest pain. Chest radiographs show mediastinal lymphadenopathy with patchy infiltrates. Most patients recover in a few weeks but some experience persistent fatigue.
Chronic Pulmonary Histoplasmosis Chronic pulmonary histoplasmosis occurs in patients with underlying lung disease and is characterized by persistent or recurrent pulmonary symptoms, progressive lung infiltrates, fibrosis, and cavitation.58,59 Upper lobe infiltrates and cavities are characteristic, resembling the findings in tuberculosis. Progression is manifested by cavity enlargement, formation of new cavities, spread to other areas of the lungs, and bronchopleural fistula. Bacterial pneumonia, Aspergillus superinfection, and malignancy also must be considered in evaluation of new masses or infiltrates.
Nodules Nodules need to be differentiated from malignancy.1,53 Calcification suggests histoplasmosis, but does not exclude malignancy. Conversely, absence of calcification, does not exclude histoplasmosis. Positive positron emission tomography (PET) scan, assumed to support malignancy, is common in histoplasmosis. Rarely, nodules may cavitate, but cavitation of nodules does not represent chronic pulmonary histoplasmosis and does not require therapy. Nodules also may rarely enlarge up to 2 mm/y, but enlargement is not caused by progressive infection and is not a basis for treatment.60 Calcification occurs in the necrotic central and surrounding fibrous tissue. An approach to the evaluation of pulmonary nodules has been reviewed.
Broncholithiasis Lymph nodes and pulmonary granuloma calcify and may erode into adjacent bronchi causing hemoptysis or obstruction.1 Patients may expectorate rock-like particles of tissue and experience recurrent and severe hemoptysis, bronchial obstruction, or tracheoesophageal fistula.
Relationship to Sarcoidosis Sarcoidosis and histoplasmosis share several clinical and radiographic findings.1 Angiotensin-converting enzyme (ACE), sedimentation rate, C-reactive protein, and immunoglobulin elevations occur in both conditions.61 Noncaseating granulomas are seen with each, and caseation, which is more typical of histoplasmosis, also occurs in sarcoidosis. Histoplasmosis is one of the most common infections that must be excluded before sarcoidosis is diagnosed to prevent mistakes in patient management, most notably administration of immunosuppressive therapy to patients with active histoplasmosis.
Mediastinal Syndromes
Mediastinal adenitis, mediastinal granuloma, and fibrosing mediastinitis are three mediastinal disorders which may complicate histoplasmosis.
Mediastinal Adenitis Mediastinal lymph node involvement is present in most cases of acute and subacute pulmonary histoplasmosis.1,53 Although usually asymptomatic, chest pain is the most common manifestation. Rarely, the airways, esophagus, or superior vena cava (SVC) may be impinged, causing obstructive symptoms. Airway obstruction is more likely in children because the airways are less rigid.
Mediastinal Granuloma Histopathology shows a necrotic central core, often containing yeast, surrounding caseating and noncaseating granuloma,1,53 fibrous tissue, and a thin-walled (less than 5 mm) capsule. The fibrosis is less exuberant than in fibrosing mediastinitis, and does not invade adjacent structures. Symptoms are usually caused by compression of the superior vena cava (SVC) esophagus, or airways, and in some cases the nodes liquefy and drain into adjacent structures.62,63 Disruption of the capsule, either spontaneous or caused by surgery, may create fistulae to the airways, pericardium, skin, or esophagus. Esophageal involvement may result in diverticulum formation.
Mediastinal Fibrosis Mediastinal fibrosis represents an exuberant scarring reaction to mediastinal histoplasmosis.1,64 The SVC is most commonly involved, but the fibrosis also may occlude airways, pulmonary arteries or veins, or esophagus, and invade the thoracic duct, recurrent laryngeal nerve, or atrium in rare cases. Chest radiographs show subcarinal or superior mediastinal widening, while CT scans reveal fibrotic restriction and invasion of mediastinal structures and calcification of the lymph nodes. Recurrent and often serious hemoptysis results from lung or airway damage and vascular compromise. Respiratory failure ensues in one-third of cases.
Other Inflammatory Syndromes
Inflammatory syndromes complicating histoplasmosis include rheumatologic syndromes and pericarditis.
Rheumatological Syndromes Patients with subacute histoplasmosis may experience arthritis or arthralgia accompanied by erythema nodosum,65 a manifestation often misdiagnosed as sarcoidosis.66 Chest radiographs usually show mediastinal lymphadenopathy and focal pulmonary infiltrates, but may be normal. These findings represent a systemic inflammatory response rather than disseminated infection, and are managed by anti-inflammatory treatment, not antifungal therapy. The illness may recur when treatment is stopped.
Pericarditis Pericarditis is another inflammatory complication of primary histoplasmosis, occurring in less than 10% of cases.67,68 Findings include chest pain, pericardial friction rub, and occasionally signs of pericardial tamponade. Chest radiographs usually show mediastinal lymphadenopathy and increase in the cardiac silhouette, while CT scan and echocardiogram may show pericardial effusion. These patients respond to anti-inflammatory treatment but may require drainage of the pericardial fluid for the management of tamponade. Late constriction is rare.
Progressive Disseminated Histoplasmosis
PDH occurs in about 1 in 2000 cases, usually in patients who are immunosuppressed or at the extremes of age. AIDS,69 solid-organ transplantation,70 and treatment with TNF-α inhibitors44 are common predisposing conditions. PDH develops during the first year of therapy in nearly half of cases, but may occur more than 10 years after transplantation. Vigilance for dissemination should be maintained throughout the course of immunosuppression.
Fever and weight loss are the most common findings in PDH.71,72 Examination reveals hepatomegaly or splenomegaly in about one-half of cases and lymphadenopathy in one-third of cases. A syndrome resembling sepsis may be seen in cases with severe immunosuppression, in whom corticosteroids are given for presumed inflammatory or autoimmune disease, or in which diagnosis is delayed. Meningitis or focal brain lesions occur in about 5% to 10% of cases.72 Other common sites of dissemination include the oral mucosa, gastrointestinal tract, skin, and adrenal glands in 5% to 10% of cases. Chest roentgenograms are abnormal in 70% of patients, usually showing diffuse interstitial or reticulonodular infiltrates, and less often a miliary pattern.71,73
In patients who experience improvement in immune function during treatment for histoplasmosis, an immune reconstitution syndrome has been reported.44,74,75 Manifestations have included respiratory failure, elevation of hepatic enzymes, hepatic or splenic abscesses, lymphadenitis, arthritis, uveitis, and intestinal obstruction.74
 DIAGNOSIS
DIAGNOSIS
Histopathology, cytology, and antigen detection are most useful for rapid diagnosis in patients with acute pulmonary and PDH, cases that require therapy (Table 134-3).22–25 A serological test for antibodies forms the basis for the diagnosis in subacute manifestations in most of the cases.24,25 Serology or culture of respiratory secretions usually provides the basis for the diagnosis of chronic pulmonary histoplasmosis. Culture results may not be available for up to 1 month, limiting its usefulness for the rapid diagnosis of severe disease.
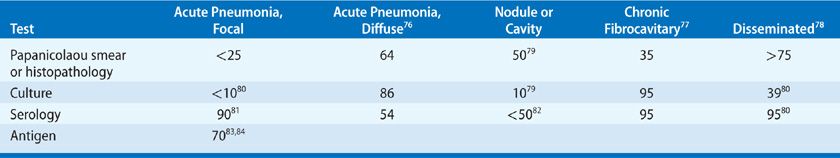
TABLE 134-3 Diagnostic Studies in Coccidioidomycosis (Percent Positive Assays)
Antigen Detection
Detection of antigen in the body fluids offers a valuable approach to rapid diagnosis in patients with PDH and acute pulmonary histoplasmosis, providing results within 24 to 48 hours.25 Antigen is found in urine of over 90% of patients with PDH and 80% with acute diffuse pulmonary disease.22,25 Sensitivity is improved by testing both serum and urine.22 Antigen detection is less sensitive in subacute pulmonary histoplasmosis.25 Detection of antigen in BAL fluid may improve the sensitivity for the diagnosis of pulmonary histoplasmosis.85 Antigen may be found in CSF in 50% of patients with Histoplasma meningitis.72 Positive results caused by cross-reacting antigens occur in patients with African histoplasmosis, blastomycosis, paracoccidioidomycosis, and Penicillium marneffei infection.25 Antigen levels decline during treatment and increase with relapse, providing a tool for monitoring therapy.86,87
Histopathology
Fungal staining permits rapid diagnosis but has a lower sensitivity than antigen detection.25 The highest yield is from bone marrow. Fungal stain of BAL is positive in 70% of cases of diffuse pulmonary histoplasmosis in patients with AIDS with PDH. The sensitivity of fungal stain of sputum or BAL in patients with other types of pulmonary histoplasmosis has not been reported but appears to be low. Yeast may be seen in peripheral blood smears in patients with severe PDH. Pneumocystis jirovecii, Candida glabrata, Blastomyces dermatitidis, C. neoformans, Toxoplasma gondii, and P. marneffei may be misidentified as H. capsulatum.
Serological Tests
Antibodies to H. capsulatum measured by immunodiffusion or complement fixation develop in most of the patients.88 Antibodies require up to 12 weeks to develop following exposure. In acute pulmonary histoplasmosis, antibodies can be detected about 15% of cases during the first 2 weeks of infection, increasing to about 75% by the sixth week.89 Elevated levels of antibodies persist for several years.90 The antibody response is greater in patients with heavy exposure and/or more severe clinical disease. Patients with asymptomatic or mild infection91 and those who are receiving immunosuppressive medications following solid-organ transplantation25 may not mount an antibody response. Antibody responses do not appear to be impaired by treatment with TNF inhibitors25,44 and are only slightly impaired in patients with AIDS.25
Serology is most useful in patients with subacute manifestations of histoplasmosis (pulmonary, rheumatological, pericarditis, mediastinal syndromes) and chronic pulmonary infection, and is positive in 90% of such cases. Sensitivity is higher while specificity is lower using complement fixation rather than immunodiffusion methods. Complement fixation titers greater than or equal to 1:32 are more suggestive of active infection but titers of 1:8 to 1:16 should not be disregarded. Cross-reactions occur in patients with other fungal diseases. Also, antibodies persisting following prior histoplasmosis may cause confusion in patients with other lung diseases.
Fungal Cultures
Cultures provide the strongest proof for histoplasmosis but are limited by low sensitivity in self-limited infections and delayed growth (2–4 weeks). Cultures are most useful in patients with chronic pulmonary histoplasmosis and PDH. Sputum production is rare in patients with acute pulmonary histoplasmosis, but organisms can be cultured from BAL or other bronchoscopy specimens in some cases. In chronic pulmonary histoplasmosis patients commonly expectorate sputum, and organisms can be found in sputum or bronchoscopy specimens in 60% to 85% of cases.25,59,85 Multiple specimens must be cultured to achieve the highest yield. In PDH, the highest yield is from bone marrow or blood, positive in over 75% of cases, and in BAL of patients with pulmonary infiltrates.85 Cultures are usually negative in pulmonary nodules and mediastinal nodes representing subacute or old healed lesions despite demonstration of yeast by histopathology.
Polymerase Chain Reaction
Several studies report diagnosis of histoplasmosis by polymerase chain reaction (PCR) in tissues or body fluids,92,93 but PCR was less sensitive than fungal stain for the examination of tissue specimens in one report.92 PCR was positive on only 8% of urine specimens with elevated Histoplasma antigen,94 22% of BAL specimens95 but not in CSF or serum.95 A role of PCR for the diagnosis of histoplasmosis remains to be determined.
 EXCLUSION OF HISTOPLASMOSIS IN WORKUP OF PATIENTS WITH COMMUNITY-ACQUIRED PNEUMONIA AND SUSPECTED SARCOIDOSIS
EXCLUSION OF HISTOPLASMOSIS IN WORKUP OF PATIENTS WITH COMMUNITY-ACQUIRED PNEUMONIA AND SUSPECTED SARCOIDOSIS
Other conditions to be distinguished from histoplasmosis include community-acquired pneumonia and sarcoidosis.
Community-Acquired Pneumonia
Histoplasmosis is always community acquired and often is initially suspected to be caused by other agents of community-acquired pneumonia.2 Failure to suspect and correctly diagnose histoplasmosis may result in mistakes in patient care, which can cause improper therapy, unnecessary morbidity or even death, and higher cost for diagnosis and management. Workup for histoplasmosis should be considered in patients from endemic areas with exposure history, if other causes cannot be established, or if empiric antibiotics do not result in prompt clinical improvement.
Sarcoidosis
Considering the similarity between the two conditions, and the risk for progression of histoplasmosis during immunosuppression for presumed sarcoidosis, active histoplasmosis must be excluded before immunosuppressant therapy is initiated.1,61 If immunosuppressive therapy was initiated in patients with laboratory findings suggestive of histoplasmosis, the patient should be followed closely for evidence of progression of histoplasmosis. Also, if patients initially respond to immunosuppressive treatment but later relapse, testing for histoplasmosis should be repeated, as immunosuppression may have accelerated the progression of histoplasmosis.
Treatment Most infections are asymptomatic or clinically self-limited, requiring no therapy. Furthermore, treatment has only been studied in PDH and chronic pulmonary histoplasmosis, precluding assessment of effectiveness in the other syndromes. While treatment in cases of acute pulmonary histoplasmosis appears to be effective, studies showing that therapy hastens the response or reduces morbidity have not been conducted. Antifungal therapy for the subacute pulmonary, inflammatory, and mediastinal syndromes is rarely indicated and probably has no effect on the course of the illness, as the pathogenesis appears to involve the inflammatory response and/or mass effects of the enlarged nodes rather than progressive infection.
Treatment guidelines have been published and should serve as reference for indications for the treatment, selection of antifungal agents, duration of therapy, and testing to monitor response.96,97 An earlier IDSA guideline summarized prior studies evaluating therapy for histoplasmosis.96
Indications for Treatment
Treatment of histoplasmosis is considered below according to the type of pulmonary involvement.
Acute Pulmonary Histoplasmosis Patients with symptomatic acute pulmonary histoplasmosis manifested by diffuse infiltrates following heavy exposure appear to benefit from antifungal therapy (Table 134-4).55,98 Adjunctive therapy with corticosteroids may hasten recovery in such patients. Amphotericin B 0.7 to 1.0 mg/kg/d would be preferred as initial therapy in patients who are more severely ill. Itraconazole, 200 mg once or twice daily, is recommended in patients with milder illnesses, and following response to amphotericin B. A 6- to 12-week course is recommended in the absence of PDH, which should be excluded.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


