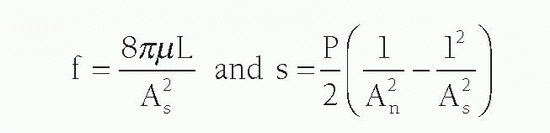The control of myocardial blood flow is based on balancing the myocardial oxygen (MVO2) supply and demand equation. The heart’s oxygen demand (through blood flow) is met through normal autoregulation by the appropriate oxygen supply. An imbalance in the oxygen demand/supply equation produces myocardial ischemia or infarction.
The heart metabolizes a variety of substrates such as glucose, free fatty acids, lactate, amino acids, and ketones, which are critical for the generation of high-energy phosphates (adenosine triphosphate [ATP] and creatine phosphate) that supply the energy requirements of the myocardium. Under normal aerobic conditions, several substrates contribute simultaneously to meeting myocardial energy needs: free fatty acid (65%), glucose (15%), lactate and pyruvate (12%), and amino acids (5%), while glycolysis plays only a minor role (
1,
2).
Determinants of MVO2 Demand
The three major physiologic determinants of MVO
2 are heart rate, myocardial contractility, and myocardial wall tension or stress (
2) (Additional factors are shown in
Table 7-1):
Heart rate is the most important determinant of MVO
2. When heart rate doubles, MVO
2 uptake approximately doubles. Increases in heart rate also increase oxygen consumption and reduce subendocardial coronary flow (
3).
Myocardial contractility is related to MVO2 consumption by the degree of pressure work per heartbeat. The decrease in MVO2 from falling ventricular wall tension is opposed by the increase in contractility. Drugs that stimulate myocardial contractility elevate MVO2 because heart size, and therefore wall tension, is not reduced substantially and is not offset by enhanced contractility.
Myocardial wall tension is proportionate to the aortic pressure and ventricular volume. Tachycardia elevates MVO2 by increasing not only the heart rate but also the frequency of tension development per unit time and the contractility.
Determinants of MVO2 Supply
MVO
2 supply depends on having an adequate inspired oxygen, normally functioning pulmonary capillary transit of oxygen, normal functioning hemoglobin to transport the oxygen and normal blood pressure, and low coronary resistance to permit blood to transit the coronary and capillary circuit. This chain of events in oxygen supply can be disrupted at any point, but insufficient supply is most commonly due to atherosclerotic obstructions.
Figure 7-1 depicts factors related to the MVO
2 supply and demand relationship.
Coronary Resistance and Flow
Coronary arterial flow is directly related to coronary resistance. Coronary resistance (R, pressure/flow) is the sum of the epicardial coronary conductance (R1), precapillary arteriolar (R2), and intramyocardial capillary (R3) resistance circuits (
Fig. 7-2) (
3). The epicardial vessels (R1) do not offer appreciable resistance to blood flow in the absence of disease. In normal arteries, there is no detectable pressure loss (i.e., resistance) along the length of the vessel (
4). The epicardial vascular smooth muscular media responds to changes in aortic pressure and modulates coronary tone in response to flow-mediated endothelium-dependent vasodilators, circulating vasoactive substances, and neurohumoral stimuli. Large conduit arteries are unaffected by myocardial metabolites because of their extramural location, but can produce episodic increases in resistance during severe focal or diffuse contraction (vasospasm) in the absence of atherosclerosis.
Precapillary arterioles (R2) connect epicardial arteries to myocardial capillaries and are the principal controllers of coronary blood flow. Precapillary arterioles (100 to 500 mm) constrict and dilate to adjust blood pressure over the range 60 to 180 mm Hg.
The microcirculatory resistance (R3) is a dense network coupling each myocyte to an adjacent capillary, and functions in a cyclic pattern with very high systolic resistance, followed by minimal diastolic resistance. Precapillary sphincters regulate flow according to oxygen demand. Several conditions, such as LV hypertrophy, myocardial ischemia, or diabetes, can impair the microcirculatory resistance (R3) and blunt the normal maximal increases in coronary flow, resulting in reduced coronary flow reserve (CFR) (i.e., the hyperemic/basal flow ratio).
Global coronary vascular resistance is regulated by several interrelated control mechanisms that include myocardial metabolism (metabolic control), endothelial (and other humoral) control, autoregulation, myogenic control, extravascular compressive forces, and neural control. These control mechanisms may be impaired in diseased states, contributing to episodic myocardial ischemia (
Table 7-2).
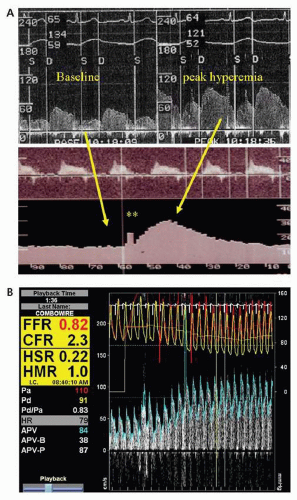
Coronary vasodilator reserve is the ability of the coronary vascular bed to increase flow from a basal level to a maximal hyperemic level in response to mechanical or pharmacologic stimuli. Such stimuli include transient coronary occlusion, exercise, or the administration of various pharmacologic agents. CFR is expressed as the ratio of maximal hyperemic flow to resting coronary flow—a ratio that averages from 4 to 7 in experimental animals, and from 2 to 5 in man (
5,
6). Factors responsible for reduced CVR in the absence of epicardial stenosis are shown in
Table 7-3.
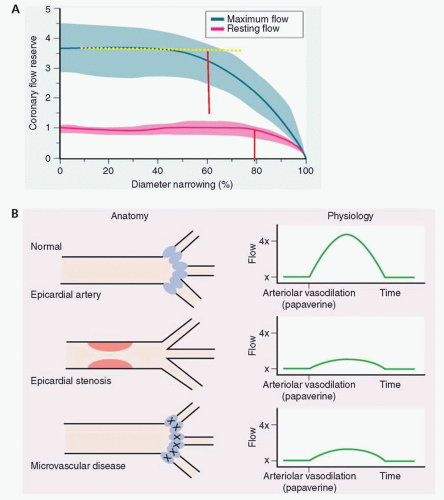
Coronary stenoses can limit coronary vasodilatory reserve (CVR). In experimental animal studies, an increasing conduit stenosis (R1) beyond 60% diameter narrowing produces a predictable decline in CVR. At diameter stenoses >80% to 90%, all available coronary reserve has been exhausted and resting flow begins to decline (
Fig. 7-3). In an individual patient, however, CVR is not useful to assess epicardial narrowings, except if normal CVR is present, indicating no significant epicardial or microvascular disease. An abnormal CVR indicates abnormality in the epicardial conduit or the microvascular resistance, or both.
Pressure Loss across a Stenosis
The resistance of a stenosis is related in large part to the morphology of the stenosis. Because resistance produces heat and the heat energy is dissipated, pressure is lost across the stenosis. As blood traverses a diseased arterial segment, turbulence, friction, and separation of laminar flow causes energy loss, resulting in a pressure gradient (ΔP) across the stenosis. The coefficients of friction (f) and separation (s) are related not only to the area of the stenosis (As) but also to the area of the normal reference segment (An) as noted here:
Where As is stenotic segment cross-sectional area, P is blood density, m is blood viscosity, L is stenosis length, and An is normal artery cross-sectional area.
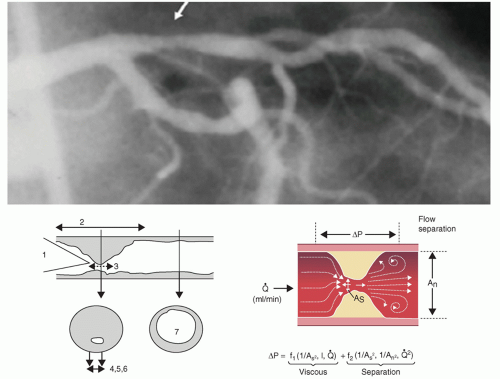
Moreover, the separation energy loss term (s) increases with the square of the flow while viscous energy loss (f) becomes negligible. Pressure loss across a stenosis thus changes exponentially with the fourth power of the radius (as assessed by the cross-sectional area) and linearly with lesion length (
Fig. 7-4). For most stenoses, the length of the narrowing has only a modest effect on its physiologic significance. However, in very long mildly narrowed segments, significant turbulence that occurs along the walls of the stenotic segment may result in a significant pressure drop. Additional factors contributing to stenosis resistance include the shape of the entrance and exit orifices, vessel stiffness, and distensibility of the diseased segment (permitting active or passive vasomotion). Dynamic changes in stenosis severity can also occur passively in response to changes in intraluminal distending pressure.