Congenital Posterolateral Diaphragmatic Hernias and Other Less Common Hernias of the Diaphragm in Infants and Children
Marleta Reynolds
The diagnosis of congenital diaphragmatic hernia can be made in utero with a great deal of accuracy. Early diagnosis allows time for parents to receive genetic and pediatric surgical consultation. Perinatal management can be altered to facilitate optimal care. The combination of early diagnosis and advances in prenatal and postnatal therapies has improved the outcome for neonates born with congenital diaphragmatic hernia. A plethora of research related to congenital diaphragmatic hernia has provided insight into the pathophysiology of this complex anomaly. Infants and older children who are found to have a congenital diaphragmatic hernia are not as severely affected as the neonate and should have 100% predicted survival.
Embryology
The classic congenital diaphragmatic hernia of Bochdalek is a posterolateral defect in the diaphragm thought to be caused by failure of the pleuroperitoneal canal to close at 8 weeks’ gestation (Fig. 53-1). Kluth and associates,31 who studied an animal model of congenital diaphragmatic hernia, believe the hernia results from defective formation of the posthepatic mesenchymal plate portion of the developing diaphragm. Babiuk and Greer,4 using animal models of congenital diaphragmatic hernia, have shown that the defect results from malformation of the amuscular mesenchymal component of the developing diaphragm rather than a defect in muscularization. They also demonstrated that the defect is a primary process and is not secondary to the development of a hypoplastic lung. The defect occurs on the left side 80% of the time and is occasionally bilateral. The hole can range in size from a 1- to 2-cm round defect to total absence of the hemidiaphragm.
When the intestines return to the abdomen from the yolk sac at 10 weeks’ gestation, the intestines and other abdominal viscera may herniate into the chest. The mediastinum is pushed into the contralateral hemithorax. Autopsy studies by the author50 and Geggel15 and their colleagues have demonstrated pulmonary hypoplasia of both lungs. The ipsilateral lung’s weight may be 20% to 50% below normal. Geggel and associates15 found the contralateral lung’s volume to be 12% to 42% below normal. The pulmonary hypoplasia is characterized by a decrease in the number of bronchioles and arterioles and in the number and size of the alveoli. Excessive muscularization of the pulmonary arterioles also occurs. Miyazaki and colleagues37 found strong insulin-like growth factor I in the hypoplastic lungs of affected neonates and proposed that the lung hypoplasia results from a failure to progress in the developing lung.
Bollmann and associates9 reported that, in 72% of fetuses diagnosed prenatally with congenital posterolateral diaphragmatic hernia, one or more extradiaphragmatic malformations were present. Eighteen percent of the fetuses studied in their group had chromosomal anomalies. Only 36.3% of the neonates diagnosed postnatally had associated malformations. In another study of fetuses with the diagnosis of congenital diaphragmatic hernia, Witters and associates64 identified associated malformations in only 26%; chromosomal anomalies were present in 9.5%. Only one infant with associated malformation survived. Congenital heart disease is the most common associated lesion and occurs in 10% to 35% of affected fetuses. When congenital heart disease is found to coexist with congenital diaphragmatic hernia, Cohen and colleagues11 found that there is a 2.9 times higher risk of death. Neural tube defects; genitourinary, skeletal, craniofacial anomalies; and abdominal wall defects can also be found. Anomalies of intestinal rotation and fixation are always present. The Congenital Diaphragmatic Hernia Study Group, as noted by Neville and coinvestigators,41 reported that, in their collected series of 1,833 patients, 23 (1.3%) had associated anomalies consistent with Fryn’s syndrome (distal digital hypoplasia, coarse facies, abnormal ears, and congenital diaphragmatic hernia). This particular subset of patients had a 17% survival rate.
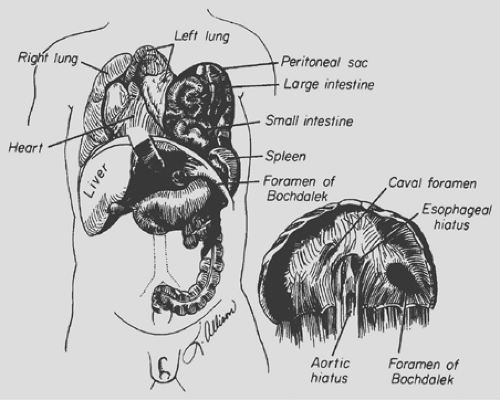 Figure 53-1. Congenital diaphragmatic hernia of Bochdalek. (From Shields TW. The diaphragm. In Nora P, ed. Operative Surgery: Principles and Techniques. Philadelphia: Lea & Febiger, 1972. With permission.) |
Attempts have been made to identify prognostic factors based on prenatal ultrasound findings. Prenatal diagnosis alone was once considered a bad prognostic factor, but as the numbers of routine ultrasounds increased, the significance of this factor decreased. Albanese and colleagues3 reviewed the records of 48 patients with prenatal diagnosis and analyzed the position of the liver on ultrasound and its relation to survival. The group with the liver in the chest32 required extracorporeal membrane oxygenation (ECMO) support more frequently (53%) than those with the liver in the abdomen (19%). Survival rate was lower in the patients with the liver in the chest (43%) than in patients with the liver in the abdomen (93%). Teixeira and associates57 reported that fetal abdominal circumference below the fifth percentile is a poor prognostic sign. Metkus and coworkers35 found that the size of the contralateral lung measured as a ratio of lung area to head circumference (LHR) was the only absolute predictor of survival. Lung-to-head ratios measured at 22 to 23 weeks’ gestation did not differ from those measured at 32 to 33 weeks and was equally predictive of subsequent survival by Jani.27 Fetal magnetic resonance imaging (MRI) has been found by Hubbard and colleagues25 to be a useful tool in confirming the diagnosis of congenital diaphragmatic hernia when ultrasound findings were inconclusive. MRI was superior to ultrasound in identifying position of the liver.
A retrospective evaluation of echocardiographic findings in fetuses with congenital diaphragmatic hernia by Sokol and associates52 found that pulmonary artery dimensions reflect lung weight. Branch pulmonary artery diameters were not hypoplastic when measured during the early midtrimester. An enlarged contralateral pulmonary artery in midtrimester correlated with postnatal death. Discrepancy in branch pulmonary artery size and a larger main pulmonary artery size correlated with postnatal respiratory morbidity. The hypoplasia of the ipsilateral pulmonary artery was progressive and may play a major role in progressive lung hypoplasia. Fuke and associates14 have demonstrated that the measurement of the ratio of acceleration time to ejection time as recorded by Doppler velocimetry was predictive of severity of pulmonary hypoplasia and survival.
Presentation
A congenital posterolateral diaphragmatic hernia may cause life-threatening respiratory distress in the first hours or days of life. The defect can cause respiratory distress or feeding intolerance in later infancy or childhood or may be identified on a radiograph obtained for unrelated reasons in an asymptomatic patient. The morbidity and mortality associated with a congenital diaphragmatic hernia are directly related to the age of the patient at presentation.
Some neonates with congenital posterolateral diaphragmatic hernia become symptomatic in the delivery room. The diagnosis is suspected if the abdomen is scaphoid and heart sounds are heard in the right chest. Radiography of the neonate demonstrates gas-filled loops of intestines in the chest (Fig. 53-2). An orogastric tube placed into the stomach to decompress the intestines may appear in the chest, and a paucity of gas exists in the abdomen (Fig. 53-3). Any neonate with respiratory distress at birth who is suspected of having a posterolateral diaphragmatic hernia should be quickly intubated. Mask bagging only increases the distention of the herniated stomach and intestines and further compromises ventilation.
Vascular access using an umbilical artery catheter is adequate for sampling arterial blood gas and the administration of fluids and drugs. The neonate should be rapidly transported to a center with a surgeon, a neonatal intensive care unit equipped to care for such an infant, and ECMO capabilities. Profound respiratory acidosis is typically found with the first arterial blood gas.
Ventilation is individualized to maintain SaO2 >90%. Prolonged hypoxemia, acidosis, and hypercarbia produce pulmonary vasoconstriction and persistent fetal circulation. Myocardial dysfunction may necessitate support with dobutamine and renal perfusion with low-dose dopamine. Because dopamine in higher doses may constrict the pulmonary vasculature, it is used only in low doses. Five percent albumin or normal saline infusions can be used to treat systemic hypotension in boluses of 10 to 15 mL.
Ventilation is individualized to maintain SaO2 >90%. Prolonged hypoxemia, acidosis, and hypercarbia produce pulmonary vasoconstriction and persistent fetal circulation. Myocardial dysfunction may necessitate support with dobutamine and renal perfusion with low-dose dopamine. Because dopamine in higher doses may constrict the pulmonary vasculature, it is used only in low doses. Five percent albumin or normal saline infusions can be used to treat systemic hypotension in boluses of 10 to 15 mL.
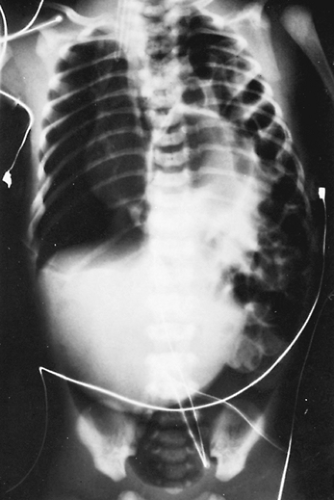 Figure 53-2. “Babygram” demonstrating multiple loops of intestine on the left side of the chest and a few loops in the abdomen. The mediastinum is shifted to the contralateral side. |
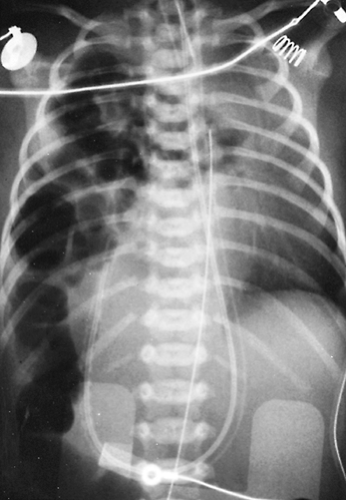 Figure 53-3. Radiograph demonstrating a right-sided diaphragmatic hernia. The orogastric tube is seen in the right side of the chest, identifying the location of the stomach. |
A congenital posterolateral diaphragmatic hernia may be found incidentally in an older infant or child. Newman and colleagues43 found that the older infant or child with a posterolateral diaphragmatic hernia may present with respiratory or gastrointestinal symptoms. Diagnosis is made with chest radiography or barium studies of the gastrointestinal tract. The hernia should be repaired at the time of diagnosis. The lungs of these children are not hypoplastic, and the operative mortality rate should be 0%.
Management
After the newborn with a congenital posterolateral diaphragmatic hernia is transferred to a center with ECMO capabilities, an effort is made to stabilize it for at least 72 hours before operative repair is considered. In the past, hyperventilation and alkalosis were used to control pulmonary hypertension and persistent fetal circulation. Wung and associates65 proposed permissive hypercapnia with spontaneous ventilation in an effort to minimize ventilator-induced lung injury (Table 53-1). Boloker and colleagues10 reported their experience in treating 120 infants using this strategy. The overall survival rate was 76%, and only 13% required ECMO.
Table 53-1 Management Strategy in Newborns With Congenital Posterolateral Diaphragmatic Hernia | ||
|---|---|---|
|
Inhaled nitric oxide (NO) is a selective pulmonary vasodilator that has been successfully used to treat pulmonary hypertension in newborns. The maximal dose recommended is 20 ppm. If persistent fetal cirulation ensues, NO should be initiated. High-frequency oscillation can also be used. In some infants, these alternative therapies will avoid the need for ECMO.
Hirschl24 described the technique of partial liquid ventilation in babies with diaphragmatic hernia. Perfluorocarbon is instilled into the trachea and lungs. Standard mechanical ventilation is performed through the liquid. Because perfluorocarbon can carry more oxygen and carbon dioxide, it can enhance gas exchange.
If these strategies are not successful in reversing persistent fetal circulation, ECMO is considered for newborns who meet institutional criteria (Fig. 53-4).
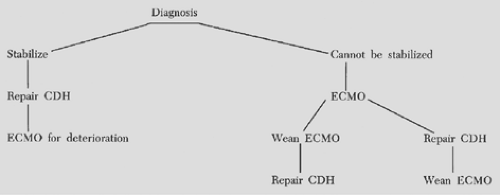 Figure 53-4. Algorithm for a symptomatic baby with congenital diaphragmatic hernia. |
Operative Correction
In the past, repair of a congenital posterolateral diaphragmatic hernia was considered a surgical emergency. Newborns were operated on in the delivery room or taken directly from the transport ambulance to the operating suite. Retrospective analysis of this treatment plan has shown little advantage or improvement in survival. In fact, Sakai and associates51 demonstrated that early operation causes deterioration in pulmonary mechanics. Nakayama and colleagues40 showed that preoperative stabilization results in an improvement in pulmonary compliance in a group of neonates with delayed repair of the hernia.This can require up to 7 to 10 days to accomplish, and no benefit has been found in repairing the hernia when the neonate is not stable and still demonstrates persistent fetal circulation.
Once the patient is stable, the neonatal ventilator is moved from the neonatal intensive care unit to the operating room for use during the operation, or the surgery is performed in the intensive care unit. Any sudden deterioration of vital signs during transport, in the operating room, or during the postoperative period usually indicates a pneumothorax on the contralateral side. Gibson and Fonkalsrud16 as well as Srouji and associates53 reported that a contralateral pneumothorax or a pneumomediastinum is associated with an increase in mortality and should be prevented with the use of low airway pressures. Needle aspiration followed by tube thoracostomy with a 10-Fr chest tube should be performed when a pneumothorax is suspected.
The correction of a congenital posterolateral diaphragmatic hernia is performed through a paramedian or subcostal incision when the defect is on the left side. For right-sided defects, a thoracic or abdominal incision can be used. The abdominal viscera are returned to the abdomen from the chest, and the hernial sac, if present, is excised. Extralobar pulmonary sequestrations, often an associated malformation in infants with congenital diaphragmatic hernia, are resected at the time of hernia repair. A small diaphragmatic defect is closed with permanent suture and Teflon pledgets. Larger defects can be closed with a polytetrafluoroethylene membrane. When the hemidiaphragm is completely absent, a polytetrafluoroethylene membrane can be sutured to the ribs, both anteriorly and posterolaterally. The medial portion of the membrane can be sutured to the contralateral diaphragmatic leaf and the adventitia overlying the aorta and esophagus. The Congenital Diaphragmatic Hernia Study Group32 reported that defect size was a major postnatal predictor of survival. Babies requiring patch repair for agenesis of the diaphragm had a survival rate of 57%; those without agenesis but needing a patch, 79%; and 95% for those not requiring a patch. The study group also reported that defect size was predictive of duration of mechanical ventilation and length of hospital stay. A chest tube may be placed in the ipsilateral thorax and attached to a three-way stopcock; it can also be used to remove fluid or air should this become necessary. Topical thrombin and cryoprecipitate are applied to the surgical field to assist in hemostasis if ECMO is anticipated. In the stable infant, the diaphragmatic defect can be closed using the minimally invasive thoracoscopic approach. Patch repair can be performed using the approach with sutures placed around the ribs if necessary.
Controversy continues regarding the best method of thoracic drainage. Suggestions have included no chest tube, bilateral prophylactic chest tubes, underwater seal, and tubes exposed to atmospheric pressure. An ipsilateral chest tube attached to a three-way stopcock allows removal of air or fluid, depending on the clinical picture and the finding on radiography of the chest. The author prefers this latter method. If fascial closure causes compromise of respiratory excursion, a ventral hernia can be created by closing the skin only. Occasionally even the skin cannot be closed, and a silo of Silastic sheeting or polytetrafluoroethylene can be used to contain the abdominal viscera temporarily.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


