CHAPTER 9 Congenital Lung Diseases
During the past decade, improvements in prenatal diagnosis1 have allowed intrauterine repair of some defects.2 With non–life-sustaining defects such as anencephaly and renal agenesis, parents may opt for pregnancy termination. For potentially correctable lesions, the pregnant mother can be transferred to a tertiary medical center. Advanced supportive and surgical techniques have led to improved survival in these infants.
HISTORY
The first report of a congenital lung defect was by Fontanus in 1639,3 describing an infant with a lung cyst. In 1777, Huber4 described an infant with an intralobar sequestration, with blood supply from the thoracic aorta. In the mid-1800s, an extralobar (Rokitansky’s lobe) sequestration was reported. The neonatal prevalence of lung infections in the 1900s was so high that it was difficult to formulate concepts of congenital lung disease, and the technology was not sufficiently advanced to correct such defects. In 1917, Gladstone and Cockayne theorized how sequestrations occurred, and in 1925, Koontz reviewed congenital pulmonary cystic diseases. The first surgical correction of a congenital lung anomaly was performed by Rienhoff5 in 1933, that being local lung cyst excision in a 3-year-old boy.6 Gross and Lewis7 performed the first pediatric lobectomy in a child with congenital lobar emphysema. Cystic disease in the right upper and right middle lobes was resected by lobectomy in 1943.8 In 1946, Gross9 performed a pneumonectomy in a 3-year-old with cystic lung disease. Potts performed the first neonatal lobectomy in 1949. Lewis later reported the death of a child on whom a lobectomy was being performed for intralobar sequestration, citing exsanguination after lost control of the systemic feeding artery. With development of advanced supportive, anesthetic, vascular, and bypass techniques, the mortality rate for children at high risk for congenital lesions has significantly improved.
EMBRYOLOGY
Lung development occurs both prenatally and postnatally and continues into adulthood. Intrauterine pulmonary development is divided into four phases: embryonic, weeks 1 to 5; pseudoglandular, weeks 5 to 16; canalicular, weeks 16 to 26; and terminal sac, weeks 26 to birth.10–12
The terminal sac period (weeks 26 to 40) is marked by continued extension of the air-blood barrier. The primitive alveoli proliferate and become intimately involved with the surrounding capillaries as the mesenchyme separating them thins out. Terminal sac formation from the terminal lung buds occurs in a coordinated interaction between promoter factors, such as fibroblast growth factor 10, and inhibitory factors, such as Sonic hedgehog and transforming growth factor β1.13,14 Mechanical fluid pressure on the terminal lung bud also appears to play a critical role.11,15,16 The adult form of alveoli has developed by the 30th to the 36th week, and the airways are nearly fully developed before term. Type II pneumocytes produce surfactant in preparation for delivery.
Normal airway and vascular development depends on a variety of factors. These include cell-to-cell interactions, local and systemic hormones and growth factors, central and peripheral neural influences, and chest wall effects. The last involve contact influences of the bony and muscular structures. Alterations in normal development may result from defects in any of these factors. Viral infection, hypoxia, starvation, and teratogens also may disrupt normal lung development. In utero airway obstruction may produce a variety of lesions.17 Eighty-four percent of these patients have additional nonpulmonary anomalies.18
Lung resection in neonates and children is fairly well tolerated. Anatomy is easily defined because there is less mediastinal and hilar fat, adenopathy, and scarring from chronic disease compared with adults. Outcomes after pediatric lung resection are good. Children younger than 5 years develop new alveoli after resection. In those older than 5 to 10 years, the alveoli enlarge, although they no longer increase in number. In an infant pneumonectomy model, lung function returns to normal within 9 to 12 months.19 Emphysematous changes do occur, as in adults. After lung resection in children in whom the remaining lung is normal, the child appears to grow, develop, and perform normally with maximal exercise.20,21 The diffusion capacity for carbon monoxide is reduced, and pulmonary artery pressure increases with exercise, but pulmonary vascular resistance is normal after resection.22
TRACHEAL ABNORMALITIES
Tracheal Agenesis and Atresia
Payne23 first described congenital absence of the trachea in 1900. There have been fewer than 100 cases reported.24 It represents a partial or complete absence of the trachea below the level of the normal larynx. This abnormality is incompatible with life if there is no connection of trachea or bronchi to the esophagus.25 Floyd and others26,27 classified three distinct types of tracheal agenesis (Fig. 9-1). In type I, the trachea originates from the esophagus, and the distal trachea, including the carina, is fairly normal; it represents 10% to 13% of the total number of tracheal agenesis cases. In type II, the trachea and carina are fused to the esophagus, and there is no residual trachea; this represents 59% to 62% of the total. In type III, the left and right main-stem bronchi originate from the esophagus, and there is no carina; this represents 22% to 31% of the total. Male incidence predominates in a 2:1 ratio. In 90% of the cases, there are other congenital lesions.28 Tracheal agenesis may be associated with VACTERL (vertebral, anal, cardiac, tracheal, esophageal, renal, and limb patterns of congenital anomalies)29 or with complex congenital heart defects as well as with upper extremity defects and duodenal atresia (referred to as TACRD).30 It is believed that tracheal agenesis is the result of developmental failure of the laryngeal tracheal bud at the third to sixth week of development. The laryngeal tracheal groove fails to develop normally.
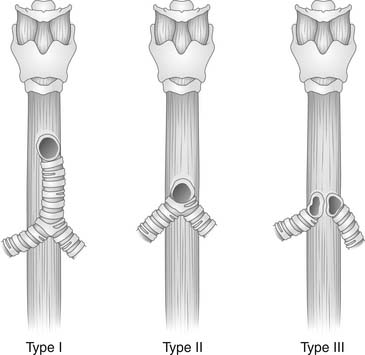
(Modified from Haben CM, Rappaport JM Clarke KD. Tracheal agenesis. J Am Coll Surg 2002;194:217-22.)
Neonates present with severe respiratory distress and the inability to cry. Survival is unlikely, although there are two reported in the literature.31,32 Those who are successfully managed are endoesophageally intubated,33 with a nasogastric tube alongside the endotracheal tube for gastric decompression. Urgent bronchoscopy or tracheostomy may assist in diagnosis.24,34 A survivor is described who had a type II defect and underwent esophageal intubation, gastrostomy, and distal esophageal banding.31 The patient is reported to have survived to 4 years, undergoing a colonic interposition at 3 years of age. A second patient is described with a type I defect who survived to 6 years of age.32 Extracorporeal membrane oxygenation (ECMO) may temporize these patients until a more definitive solution can be achieved.35 If the patients can be stabilized, they should be examined for other life-threatening defects, such as neural and cardiac anomalies, that may require correction. The gastroesophageal continuity can be corrected with colonic interposition or gastric conduit, but there have been no attempts to reconstruct the trachea. Perhaps, for the future, to reconstruct the incompletely developed airway, tracheal allografts36 or tissue-engineered replacements37 may be used for long-term success.
Congenital Tracheal Stenosis
Congenital tracheal stenosis (Fig. 9-2) is rare. Cantrell and Guild38 described three forms of stenosis. Type I is stenosis of the full length of the trachea. Type II is a funnel-shaped stenosis of the upper, lower, or entire trachea. Type III is a segmental stenosis of the lower trachea. This classification assists in identifying patients who are more likely to have associated anomalies and in planning treatment. Stenotic regions have complete cartilaginous rings, the number of which is variable, anywhere from 2 to 18. This rare anomaly is associated with pulmonary vascular sling or vascular ring in 50% of cases.39 A functional classification system has been proposed to assist in selection of the operative candidate.40,41
These patients usually have exertional wheezing or stridor after birth. The type II and type III abnormalities are associated with anomalous location of one more bronchi, most likely a left pulmonary artery sling. All three types are associated with pulmonary agenesis. Symptoms vary according to the degree of stenosis. Patients presenting during the neonatal period or infancy with severe stridor, cyanosis, and recurrent stridor have the highest mortality. Many of these patients have associated congenital malformations and infections. Their prognosis even after surgical correction is variable.40 Other children presenting at a year of age or soon thereafter have a surgical mortality of 10% to 20%.42 Some children, late in childhood or early teens, may present for an evaluation for asthma with varying degrees of exertional dyspnea. Typically, these children have a better prognosis. CT, especially multiple detector CT, provides enough detail to assess the tracheobronchial abnormality with a sensitivity of 90%, better than flexible bronchoscopy,43 and any associated anomalies. Bronchoscopy aids in diagnosis, treatment planning, and evaluation of the surgical repair intraoperatively and postoperatively. Echocardiography is helpful to assess for cardiac and vascular anomalies. Patients should be aggressively treated because they are at high risk for sudden death without surgical correction.44 Partial tracheal resection is the most frequently performed operation. The neonatal trachea does not tolerate tension as well as in the adult.45 Up to 50% of the infant trachea can be resected with primary reanastomosis. The repair is much more difficult if more than 50% of the trachea or the carina is involved.
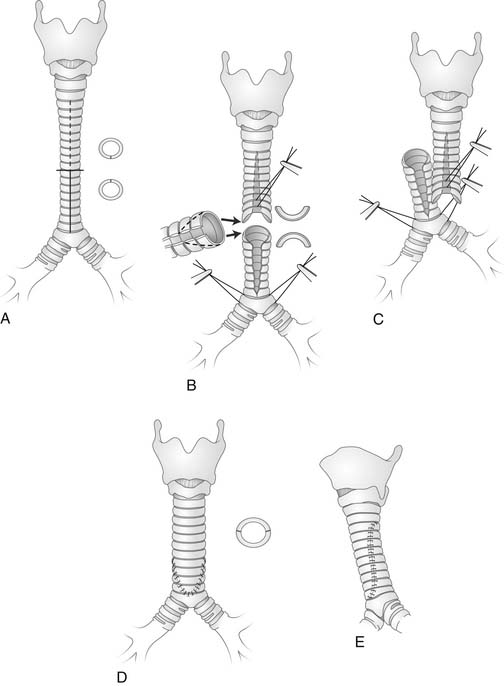
In 1964, the first repair, a primary resection and end-to-end anastomosis, was reported by Cantrell and Guild.38 Reconstruction has been performed with pericardium,46 aortic homograft,47 costochondral graft,48 slide tracheoplasty (Fig. 9-3),49,50 and tracheal autograft.51 Complete repair of the tracheal stenosis and any vascular ring or sling requires cardiopulmonary bypass and potential circulatory arrest. Backer and coworkers52 reviewed their 18-year experience in 50 patients, comparing four tracheal stenosis repair techniques: pericardial patch, primary resection, tracheal autograft, and slide tracheoplasty. They concluded that when eight or fewer rings are involved, primary resection is preferable. For stenosis longer than eight rings, an autograft is better. Vascular endothelial growth factor application has the potential of improving the results of tracheal autograft.53 In contrast, Muraji54 demonstrated that the slide tracheoplasty technique allowed early extubation and an excellent long-term result in one case. ECMO may be necessary to support the neonate postoperatively.35,48 Long-term outcome after resection appears to result in normal growth and development.55 For patients who are considered surgically uncorrectable, palliative maneuvers include balloon dilation and split posterior tracheoplasty,56,57 stenting, local steroid injection, electroresection, and cryotherapy.40,41
Tracheomalacia
In tracheomalacia, weakened cartilage collapses on expiration. With inspiration, there is sufficient suspension of the tracheal wall to maintain patency. The most common pathologic finding is oval rather than round cartilaginous rings. There is a congenital and an acquired form of tracheomalacia. The congenital form can be diffuse and both proximal and distal, involving the main-stem bronchi. It is associated with tracheoesophageal fistula.58,59 The more common acquired form results from degeneration of the cartilaginous support, usually by vascular compression such as from a vascular ring or sling, adjacent inflammatory process, tumor, or chest wall deformity (e.g., pectus excavatum).
One fourth of all esophageal atresia patients have tracheomalacia. Tracheoscopy, bronchoscopy, esophagoscopy, and esophagography may be necessary for complete evaluation. CT and MRI provide additional information. The differential includes normal pulsatile collapse, bronchial webs, and tracheal tumors. Many patients suspected of having tracheomalacia may instead have gastroesophageal reflux disease or reflux disease in addition to tracheomalacia. This may warrant either a trial of a proton pump inhibitor and prokinetic agent or a formal evaluation for reflux.60
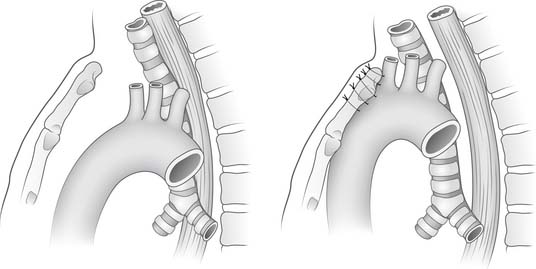
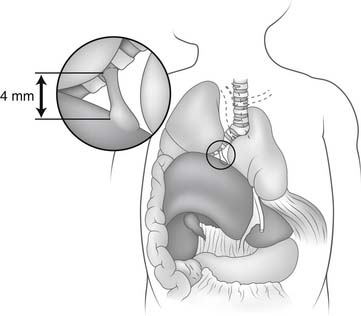
Tracheomalacia improves with age in most cases. “Dying spells,” inability to be weaned from the ventilator, and failure to thrive are indications for more aggressive therapy. If tracheomalacia is mild, the infant should be supported and observed because infants usually outgrow their tracheomalacia within the next 2 years. In more severe forms, mortality is as high as 80% with conservative management. Stents have been used with varying success to allow cartilage maturation, usually taking several months.61,62 Tracheostomy also has been used but is associated with some complications.63 Aortopexy (Fig. 9-4), by itself or with tracheal resection or stenting, has been performed by a left submammary thoracotomy through the third interspace. The thymus is preserved. After exposure of the ascending aorta, three or four nonabsorbable sutures are placed through the aortic adventitia above and below the innominate artery takeoff. These sutures are attached to the periosteum. Finally, the endotracheal result is visualized by intraoperative bronchoscopy. Eighty percent of the patients are extubated by 4 weeks, and the long-term results are good.64 The acquired form is treated similarly to the congenital form. Longer segment lesions without vascular compression often require tracheostomy and mechanical ventilation for a time. Bronchomalacia may require both aortopexy and pulmonary artery pexy.65
BRONCHIAL BRANCHING ABNORMALITIES
Bronchial branching abnormalities are rarely symptomatic. Accessory bronchi can originate off the trachea or main-stem bronchi and may be attached to rudimentary lung. They are usually incidental findings at autopsy. Cervical diverticula present in utero may regress before birth. There are case reports of bronchi crossing the mediastinum to supply the opposite lung.66 The most frequent tracheal bronchus is to the right upper lobe. These abnormalities may be isolated or associated with other tracheopulmonary anomalies. They can be associated with wheezing, stridor, pneumonia, bronchiectasis, and hemoptysis. Accessory bronchi and associated abnormal pulmonary tissue should be resected if symptomatic.67 Chest radiography and high-resolution CT are helpful in defining the anomaly.68
Bronchial Atresia
Bronchial atresia, first described by Ramsay in 1953,69 is the second most common abnormality of the airway after tracheoesophageal fistula. A lobar or segmental bronchus ends blindly in the lung tissue. Lung tissue distal to the bronchial atresia expands and becomes emphysematous as a result of air entering through the pores of Kohn.70 Beyond the atretic segment but proximal to the hyperinflated lung, the terminal airway is mucus filled. It is believed that the bronchial bud somehow separates distally from the proximal bronchial bud and continues to develop. Another potential explanation is that there is a vascular insult of the airway in the atretic segment. In either case, the distal airway continues to develop normally.
For better assessment of this abnormality, a CT scan should be performed.71,72 Prenatal diagnosis by ultrasonography has been reported.73 The differential diagnosis includes acquired bronchial stenosis (which may result from long-term intubation), bronchial adenoma, sequestration, mucoid impaction, vascular compression syndrome, Swyer-James syndrome, and atypical bronchogenic cyst.
Indications for resection in bronchial atresia include a recurrent and possibly serious pulmonary infection, respiratory distress, and increasing size of the translucent lung.74 Some resect the abnormal tissue to prevent infection. Segmentectomy is possible, but lobectomy is usually required.
Congenital Bronchiectasis
Bronchiectasis is an abnormal dilation of the bronchi or bronchioles. It is believed to be secondary to failure of the mesenchyme to differentiate into cartilage and muscle.75 It results in a chronic, mildly productive cough with recurrent pneumonia. CT usually achieves the diagnosis,76 and bronchoscopy typically is unnecessary. Bronchography also is unnecessary, given the detail provided by CT. Treatment and prognosis depend on the number of segments or lobes involved. Patients are provided with humidity, chest physiotherapy, and oxygen as necessary. Therapeutic bronchoscopy is performed for suspected mucoid obstruction or bleeding. Bleeding is initially controlled by embolization, topical epinephrine, or intravenous pitressin. Oral, intravenous, and nebulized antibiotics may be helpful in controlling symptoms.77 Patients who have localized congenital symptomatic bronchiectasis may require a segmentectomy, lobectomy, or possibly pneumonectomy. The success of surgical intervention is related to the ability to resect all the diseased lung.78
Tracheobronchomegaly (Mounier-Kuhn Syndrome)
Tracheobronchomegaly was first described in 1932 by Mounier-Kuhn.79 It is characterized by excessive dilation of the trachea and main bronchi (Fig. 9-5).80 This dilation is the result of atrophy of the elastic tissue and smooth muscle of the trachea and main-stem airways.81 Airway dilation continues to third-order subdivisions. During the course of the disease, mucosal herniations develop between tracheal rings, creating a diverticulosis pattern. Associated conditions include cutis laxa, Ehlers-Danlos syndrome, Marfan syndrome, Kenny-Caffey syndrome, ataxia-telangiectasia, ankylosing spondylitis, Brachmann–de Lange syndrome, and light chain deposition disease. When an isolated process occurs, there is recessive inheritance.82 Tracheobronchomegaly also may result from severe pulmonary fibrosis with increased traction on the tracheal wall.83,84 As these diverticula develop, there is mucus retention with resultant development of bronchiectasis and fibrosis. Patients eventually are unable to clear secretions and succumb to chronic infection and respiratory failure.
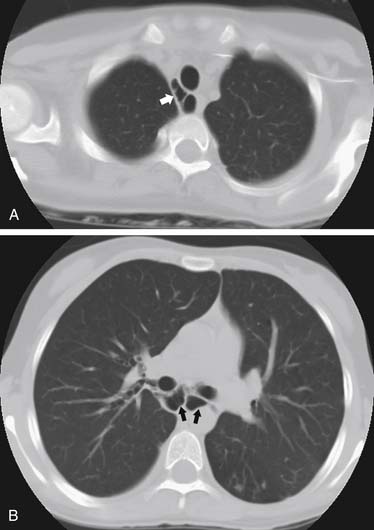
The patients are usually men in their third to fourth decades of life. Fifty percent of patients with tracheobronchomegaly have no symptoms until their third decade. They usually have recurrent lower respiratory tract infections. Asymptomatic patients may be identified in that the trachea is three standard deviations larger than normal; for adults, it exceeds 3 cm.85 Patients with tracheobronchomegaly frequently have recurrent respiratory tract infections and persistent cough. Plain chest radiography demonstrates an enlarged trachea and main-stem airways that may be ectatic. CT more clearly demonstrates changes, and tracheal dimensions for adults86 and children87 are helpful in making the diagnosis. Bronchography may be necessary to differentiate it from acquired bronchiectasis. Bronchoscopy generally is unrewarding, with inadequate visualization of the airway lumen as a result of multiple tracheal wall diverticula.
In general, there is no surgical role in the treatment of tracheobronchomegaly. Management of secretions helps minimize the symptoms. Stenting and T-tubes88,89 may provide significant palliation. Recently, a successful double-lung transplantation with bilateral bronchial stents for tracheobronchomegaly has been reported.90
Laryngotracheoesophageal Cleft
Laryngotracheoesophageal cleft is a rare congenital abnormality resulting from failure of the esophagus to separate from the laryngotrachea91; infants have a toneless cry and choke with feeding. On an attempt to pass a nasogastric tube, a communication with the airway is seen. A barium study or endoscopy can demonstrate the fistula. One half to more than three fourths of patients survive.92,93 With late detection, mortality is high. Patients are first treated with a gastrostomy. Then, through a cervical approach, the fistula is divided, the esophagus and larynx are repaired, and a muscle flap is placed between them to reduce likelihood for recurrence. A nasogastric stent is left in place for a time. For large defects, a combined cervical and thoracic approach may be necessary.94
Tracheobronchial-Esophageal Fistula
Tracheobronchial-esophageal fistula (TEF) is the most common abnormality of the trachea, occurring in 2.4 per 10,000 births.95 The most commonly used anatomic classification system for TEF is the one proposed by Gross.96 type A, esophageal atresia without TEF; type B, esophageal atresia with proximal TEF; type C, atresia with distal TEF; type D, atresia with both proximal and distal TEF; and type E, TEF without esophageal atresia (H-type). Type C is the most common, accounting for 87% of the anomalies, followed by type A (8%), type E (4%), type B (1%), and type D (1%). Associated congenital defects are common97; cardiovascular anomalies lead the list, with concomitant occurrence in 35% of TEF cases. In about 20% of cases, TEF occurs as part of the VACTERL constellation of defects.
Patients present with feeding difficulties and excessive salivation. Intermittent cyanosis and tracheobronchial infections are seen as well. Diagnosis of the most common form (type C) is suspected when a large amount of air is seen in the esophagus, stomach, and small bowel. Failure of nasogastric tube passage and its position on plain radiography or contrast fluoroscopy confirm the condition. Cameron Haight is credited with the first successful pediatric TEF repair.98 Although it was initially accomplished through left thoracotomy, Haight came to prefer right extrapleural thoracotomy for division of the fistula and esophageal anastomosis. Esophageal anastomotic leak is seen in about 15% of cases, but with a functioning chest tube in proximity, 95% of leaks heal spontaneously.99 Subsequent stricture formation is common, but it generally responds well to dilation. Major, life-threatening leaks occasionally occur. In this situation or in the case of so-called long-gap atresia, wherein esophageal continuity cannot be established despite a variety of elongation techniques, esophageal replacement by colonic interposition or gastric transposition may be necessary. Even in critically ill neonates with a substantial esophageal anastomotic leak, gastric pull-up has been successfully applied as an alternative to temporizing with wide drainage alone.100 Long-term outcome after surgery for TEF has been the subject of several recent reviews.101–103
For ligation of the H-type fistula, a right neck incision is made, allowing avoidance of the thoracic duct. The sternocleidomastoid muscle and carotid sheath contents are retracted laterally. The recurrent laryngeal nerve is identified and protected, and the fistula is divided and oversewn. An interposition muscle flap reduces the likelihood of recurrence. TEF recurrence has been successfully managed with a minimally invasive technique. In one pediatric series, the fistula tracts were de-epithelialized by fulgurating diathermy under endoscopic guidance, with successful track obliteration by way of fibrin glue application.104 For the lower-lying H-type fistula not accessible through a cervical incision, thoracoscopic ligation has been successful.105,106
Not surprisingly, minimally invasive techniques have been increasingly applied not only to management of fistula obliteration but to esophageal reconstruction as well.107–109 Thoracoscopic repair has been accomplished through an extrapleural approach,110 and even long-gap esophageal atresia has been amenable to the thoracoscopic approach. The latter consists of initial thoracoscopically mediated esophageal traction followed by staged thoracoscopic creation of the esophageal anastomosis.111 Nuances of anesthetic and intensive management of neonates undergoing thoracoscopy and laparoscopy are becoming better appreciated.112,113 Meticulous anesthetic management has allowed successful TEF repair in a neonate with single-ventricle physiology (unpalliated tricuspid atresia).114
Preservation of the azygos vein, previously divided with impunity during repairs of esophageal atresia and TEF, has been advanced as desirable. The theory is that postoperative mediastinal edema may be avoided, thereby minimizing esophageal anastomotic complications.115,116 Clinical significance of this concept awaits further evaluation.
Bronchobiliary Fistula
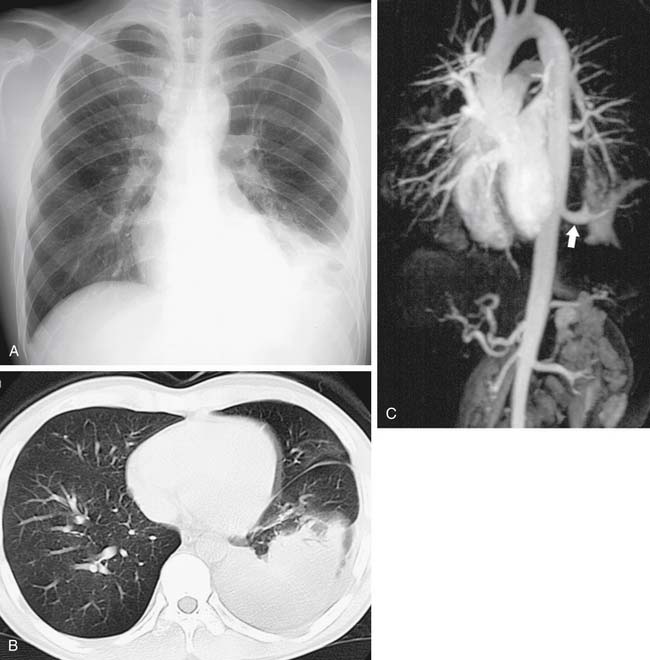
Bronchobiliary fistula (Fig. 9-6) is a very rare congenital abnormality with perhaps fewer than 50 cases reported.117 It has generally been reported in infants and children, but a number of cases have been diagnosed in adulthood.118–120 The fistula track is composed of bronchial and biliary-type tissue from its origin in the airway to the liver. Patients produce green sputum and develop dyspnea and chronic cough. Infants may present with respiratory distress121; girls are more commonly affected. The right middle lobe and right main-stem airways are most frequently involved and usually drain to the left hepatic ductal system. Associated anomalies, although not the rule, include esophageal atresia and TEF,122 biliary atresia,123 and right-sided diaphragmatic hernia.117
Diagnosis is made by bronchoscopy, during which a catheter may be passed into the fistula for administration of contrast material and concomitant radiography.124 This represents a more modern form of bronchography. Diagnosis also has been achieved with a nuclear biliary scan (99mTc-HIDA cholescintigraphy)125,126; this is useful for postoperative surveillance as well. Finally, MRI for neonatal diagnosis has been reported, emphasizing the utility of T1-weighted gradient-echo sequences.127
Patients with a confirmed bronchobiliary fistula are treated by ligation and division of the fistula, transecting it as close to the airway as possible. Pulmonary resection has been necessary on occasion.119 Access is by way of transpleural or extrapleural right thoracotomy. Cholecystography is used selectively to verify biliary drainage into the duodenum.126 For those with impaired prograde biliary drainage or biliary sepsis, hepatic lobectomy (generally left-sided) or Roux-en-Y hepaticojejunostomy may be necessary.128
Sequestrations
Sequestrations are lung defects that have systematic arterial blood supply and abnormal bronchial connections. They are invariably located in the base of the lung, with males more frequently affected than females by a 3:1 ratio. Intralobar sequestrations (Fig. 9-7) are cystic abnormalities within the visceral pleural covering of the lung, whereas extralobar sequestrations (Rokitansky’s lobe; Fig. 9-8) are mass lesions that have their own separate visceral covering. Intralobar sequestrations represent 75% of all sequestrations. As a group, intralobar sequestrations receive vascular blood supply from the thoracic aorta in 74% of cases, from the abdominal aorta in 19% of cases, from the intercostals in 3% of cases, and from multiple sources in 20% of cases. The aberrant blood supply most commonly enters the anomalous lung away from the hilum. The venous drainage of sequestrations is usually through pulmonary veins, but it may be through systemic veins. Intralobar sequestration is most frequently found within the posterior segment of the left lower lobe.
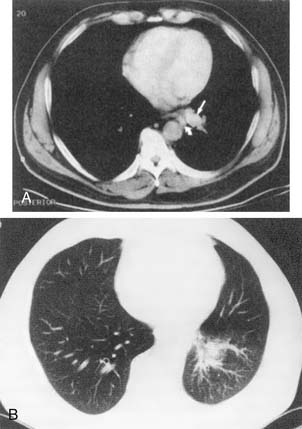
Intralobar and extralobar sequestrations are both derived from foregut tissue and thus may possess an esophageal fistula. Intralobar sequestrations may present in utero as polyhydramnios or may become symptomatic in adolescents or young adults. Repeated bouts of pulmonary infection and hemoptysis are the most common symptoms. Intralobar sequestration is rarely associated with other congenital anomalies; extralobar sequestration is not uncommonly associated with other anomalies, especially congenital diaphragmatic hernias. Extralobar sequestrations may be found within either the pericardium or the diaphragm or beneath the diaphragm retroperitoneally. Malignant neoplasms have been reported within extralobar sequestrations, and a simultaneous discovery of congenital cystic adenomatoid malformation has been reported, as have other anomalies such as pericardial cysts, esophageal achalasia, and cardiac defects. In the absence of malignant disease, patients with both intralobar and extralobar sequestrations may demonstrate marked serum elevations of the tumor markers CEA and CA19-9.129
CT is sensitive and specific for identification of either intralobar or extralobar sequestrations. MRI may be helpful.130 Bronchography and bronchoscopy are less likely to provide additional information. Barium esophagography may be necessary to demonstrate an esophageal communication. Angiography is seldom necessary, because noninvasive studies generally demonstrate the feeding systemic vessels.
Many lesions are discovered prenatally, and careful follow-up is recommended. If hydropic changes are noted, delivery is indicated.131,132 In infants, cysts that are large or cause hemodynamic compromise may be temporarily drained by needle aspiration or tube thoracostomy to allow adequate induction of anesthesia. If no infection is involved, the cyst alone, or possibly a segment, may be removed. In most cases, however, lobectomy is required. Early identification of the aberrant arterial blood supply is critical and helps avoid disastrous bleeding. Most of the arteries are less than 1 mm in diameter, but some have been reported to be as large as 2.5 mm. Atherosclerotic changes in the systemic artery may make it difficult to manipulate during surgery. A communication with the gastrointestinal tract should be thoroughly searched for at the time of surgery. Venous drainage also may be aberrant, and it should be ascertained before resection, if possible. Both intralobar and extralobar sequestrations may be amenable to resection by minimally invasive techniques. The outcomes after resection are excellent.133,134
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


