Congenital Lesions of the Lung
Marleta Reynolds
Most congenital lesions of the lung are recognized when respiratory symptoms develop in the newborn or infant. An increasing number are being diagnosed prenatally. Some are identified in the asymptomatic child on an incidental radiograph of the chest. The remainder are diagnosed in the older child during evaluation for a respiratory infection. In an infant with severe respiratory distress, knowledge of the pathology and the ability to make a quick and accurate diagnosis based on a radiograph of the chest may be critical. Ultrasonography and computed tomography (CT) allow greater diagnostic accuracy when time permits.
Tracheal Agenesis and Atresia
Tracheal agenesis or atresia leads to respiratory distress at birth and is usually fatal. Floyd and colleagues20 categorized the anomalies into three subtypes. Type I (10%) includes partial atresia of the trachea with a normal short segment of distal trachea arising from the anterior esophageal wall. Type II (59%) is complete tracheal agenesis with normal bronchi, bifurcation, and carina, with the carina connecting to the esophagus. Type III (31%) is complete agenesis of the trachea, with the bronchi arising from the esophagus. Affected babies may be premature, and maternal polyhydramnios is often associated. At birth, the babies turn blue and do not have an audible cry. An endotracheal tube cannot pass beyond the vocal cords, but esophageal intubation may temporarily improve ventilation. Associated anomalies are present in 84% and include other bronchopulmonary malformations, cardiac defects, vertebral anomalies, and gastrointestinal anomalies, as reported by Manschot and associates.45 Hiyama and colleagues29 described a baby with tracheal atresia who was successfully managed with multiple surgical procedures. The first procedure for the type II lesion included banding of the distal esophagus and gastrostomy. An endotracheal tube was then passed into the esophagus to just above the bronchoesophageal fistula. After 9 months of mechanical ventilation and gastrostomy feedings, a tracheostomy was created into the upper trachea. The esophagus was disconnected from the trachea and later reconstructed with a colon interposition. At the time of the report, the child was 4 years old. Sandu and Monnier61 refer to three other survivors treated with multiple and varied surgical procedures.
In 2000, Crombleholme and associates14 described the first survivor with complete high airway obstruction syndrome (CHAOS) successfully treated with an ex utero intrapartum treatment (EXIT) procedure.
Bronchial Anomalies
Abnormal bronchial development may result in complete bronchial atresia, an aberrant origin of a mainstem or segmental bronchus from the trachea, or the abnormal communication of the bronchus with another foregut derivative. Structural abnormalities of a bronchus may lead to lobar emphysema (Fig. 83-1).
Tracheal Diverticulum and Bronchus
A tracheal diverticulum may arise from the cervical or thoracic portion of the trachea and ends blindly or in a rudimentary lung. Early and Bothwell17 reported two infants in whom a tracheal diverticulum was discovered on bronchoscopy. Computed tomography (CT) in conjunction with bronchoscopy can correctly identify the lesion. Surgical resection is reserved for those in whom the diverticulum is large enough to cause compression of adjacent structures or becomes a source of continued pulmonary sepsis.
If the bronchial structure connects to a normal segment or lobe of lung, it is referred to as a tracheal bronchus. The accessory lung has normal pulmonary arterial and venous supply. Symptoms may result from stenosis of the tracheal bronchus or from other associated pulmonary anomalies. A radiograph of the chest obtained because of recurrent pneumonia, stridor, or newborn respiratory distress reveals the portion of lung involved (Fig. 83-2). At bronchoscopy, the tracheal bronchus can be seen, and an assessment of the entire tracheobronchial tree should be performed. Conventional CT and high-resolution CT scanning can be used to demonstrate a tracheal bronchus. Wong and colleagues78 recommend serial coronal CT scans to better demonstrate a tracheal bronchus. Bronchography is seldom needed. Surgical resection of the tracheal bronchus and adjoining lung tissue is necessary only when it is clinically indicated (Fig. 83-3). McLaughlin and colleagues49 recorded 18 children with tracheal bronchi who presented with recurrent pneumonia and other respiratory symptoms. Bronchography revealed bronchial stenosis and bronchiectasis of the involved lung segment in five of the children. Their symptoms were relieved by resection. Vevecka and colleagues73 reported a 1-year-old child who was found to have a cystic adenomatoid malformation connecting to a tracheal bronchus.
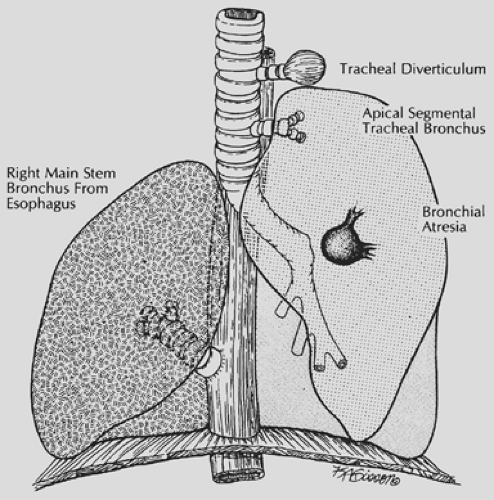 Figure 83-1. Bronchial anomalies resulting from abnormal bronchial development. Symptoms result from bronchial obstruction and pulmonary infection. (From Luck SR, et al. Congenital bronchopulmonary malformations. Curr Probl Surg 1986;23:251. With permission.) |
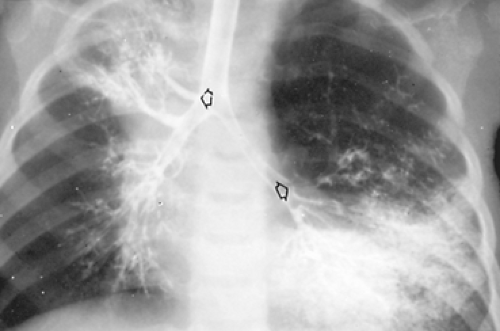 Figure 83-2. Incidental tracheal bronchus. The patient, a 3-year-old with a history of wheezing, also had an obstructed left-upper-lobe apical bronchus and emphysema of the left upper lobe. Left upper lobectomy was curative. (From: Raffensperger JG. Congenital malformations of the lung. In Raffensperger JG, ed. Swenson’s Pediatric Surgery, 4th ed. Norwalk, CT: Appleton-Century-Crofts, 1980:700. With permission.) |
Bronchial Atresia
An atretic bronchus ends blindly in lung tissue. At birth, the portion of lung adjacent to the atretic bronchus is filled with fluid. The fluid is soon reabsorbed and replaced by air from the adjacent lung tissue through the pores of Kohn. Eventually, retained secretions result in a mucocele. Compression of adjacent normal bronchial structures leads to emphysematous change in the lung. Symptoms of wheezing and stridor may develop, and there is a significant risk for pulmonary infection. A radiograph of the chest shows a hilar mass with radiating solid channels surrounded by hyperaerated lung. CT scanning can differentiate the centrally placed cystic mucocele characteristic of bronchial atresia from a bronchogenic cyst or lobar emphysema. Resection is indicated to prevent pulmonary sepsis. For 9 years, Haller and colleagues23 observed a child with mild symptoms who had bronchial atresia and associated lobar emphysema. Progressive respiratory symptoms eventually prompted resection of the involved segment. The report documents the natural history of the lesion and further supports timely resection. Morikawa and associates51 reported 29 children with bronchial atresia ages 1 day to 13 years. All but one were symptomatic and treatment with lobectomy or segmentectomy was successful.
Kunisaki and associates36 evaluated the surgical specimens from 26 lung resections performed to treat prenatally
diagnosed lung lesions. Bronchial atresia was found in 77% of the specimens, suggesting that this entity is associated with multiple congenital lung malformations. Riedlinger and colleagues57 expanded on this group of patients and described atresia of a lobar, segmental, or subsegmental bronchus in 100% of cases of extralobar sequestration, 82% of intralobar sequestration, 70% of congenital cystic adenomatoid malformation (CCAM), and 50% of lobar emphysema. Keswani and associates33 reported two cases of atresia of the mainstem bronchus. Fetal hydrops developed as the associated lung grew rapidly in size. A fetal pneumonectomy was performed in one fetus, but it expired during the procedure. In the other case, mirror syndrome developed in the mother and labor was induced. A nonviable fetus was delivered at 25 weeks.
diagnosed lung lesions. Bronchial atresia was found in 77% of the specimens, suggesting that this entity is associated with multiple congenital lung malformations. Riedlinger and colleagues57 expanded on this group of patients and described atresia of a lobar, segmental, or subsegmental bronchus in 100% of cases of extralobar sequestration, 82% of intralobar sequestration, 70% of congenital cystic adenomatoid malformation (CCAM), and 50% of lobar emphysema. Keswani and associates33 reported two cases of atresia of the mainstem bronchus. Fetal hydrops developed as the associated lung grew rapidly in size. A fetal pneumonectomy was performed in one fetus, but it expired during the procedure. In the other case, mirror syndrome developed in the mother and labor was induced. A nonviable fetus was delivered at 25 weeks.
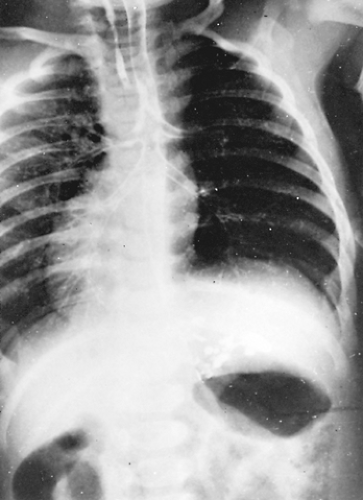 Figure 83-3. Bilateral tracheal bronchi in a child being evaluated for chronic cough. Conservative treatment was recommended. (From Holinger PH. Abnormalities of the larynx and tracheobronchial tree. In Swenson O, ed. Swenson’s Pediatric Surgery, 3rd ed. Norwalk, CT: Appleton-Century-Crofts, 1969:297. With permission.) |
Anomalous Bronchi
The most common communication between the trachea or bronchus and another foregut derivative is a tracheoesophageal fistula. Other anomalous connections are rare. Since 1950, Gans and Potts21 and Nikaido and Swenson53 have reported three infants from my institution who had an esophageal bronchus. All presented with respiratory distress and pneumonia and were found by esophagogram to have a portion of lung communicating with the esophagus through a bronchial structure.
The anomalous origin of a lobar or segmental bronchus from the esophagus (esophageal bronchus) may be right- or left-sided and may involve the upper or lower lobes. The vascular supply to the involved lobe varies; some lobes have a normal pulmonary vascular pattern and others a systemic supply. Extralobar and intralobar sequestrations (identified by the anomalous blood supply and absence of bronchial communication) occasionally communicate with the esophagus or other foregut derivatives. Confusion over terminology has prompted some researchers to refer to all these anomalies as congenital bronchopulmonary foregut malformations.
Persistent or recurrent pneumonia in an infant or child may be caused by bronchial communication with the esophagus. An esophagogram outlines the esophageal bronchus (Fig. 83-4). Resection of the chronically infected portion of the lung is indicated. Lallemand and associates38 reported three cases of esophageal bronchi. In two cases, the anomalous bronchus was a mainstem bronchus. These two patients were treated by implantation of the anomalous bronchus into the trachea. Linnane and Canny40 report a similar patient who presented with respiratory distress at 5 months of age. At surgery, the right mainstem bronchus ended abruptly. The existing right lung communicated with the lower third of the esophagus and had a systemic arterial and venous blood supply. The sequestered lung and communication with the esophagus was successfully resected.
Bronchial Stenosis
A true congenital bronchial stenosis is rare. Bronchial stenosis is seen most often in the right mainstem bronchus secondary to inflammatory changes after improper and frequent suctioning of an infant on prolonged ventilatory support. Granulation tissue builds up at the mainstem orifice. The lung distal to the obstruction becomes chronically infected or emphysematous (Fig. 83-5). Repeated bronchial dilations may be necessary to restore normal lung function and treat the infection. Jaffe32 reported successful balloon dilation in four of six patients with congenital and acquired tracheal and bronchial stenosis. Bronchoplastic procedures may reduce the need for pulmonary resection when the chronic infection does not resolve after attempts at bronchial dilation.
Tracheobiliary and Bronchobiliary Fistula
Only 20 cases of tracheobiliary and bronchobiliary fistula have been reported. Affected infants present with mild or severe respiratory distress and may have bile-stained secretions. Recurrent pneumonias may prompt a chest radiograph. Bronchoscopy and fistulography are diagnostic. Egrari and associates18 used a hepatoiminodiacetic acid scan to demonstrate the anomaly in an affected infant. The fistula usually joins the right mainstem bronchus near the carina. Division of the fistula is usually curative. Ferkol and colleagues19 described an infant with bronchobiliary fistula. The biliary drainage of the left lobe liver was through the fistulous tract in this patient. Division of the fistula was followed several days later by resection of the fistulous tract and the abnormal left lobe of the liver because of biliary sepsis. In the review of this type of malformation by Tommasoni and associates,72 the fistulas were reported to have arisen from
the carina in 55%, the right mainstem bronchus in 30%, the left bronchus in 10%, and was unidentified in 5%.
the carina in 55%, the right mainstem bronchus in 30%, the left bronchus in 10%, and was unidentified in 5%.
The fistulas end in the left hepatic biliary ductal system. Associated biliary anomalies are present in 56%. Identification of a normal biliary tree and continuity with the duodenum is mandatory. The fistula may provide a substantial portion of the biliary drainage of the liver and a fistula–enteric anastomosis may have to be constructed.
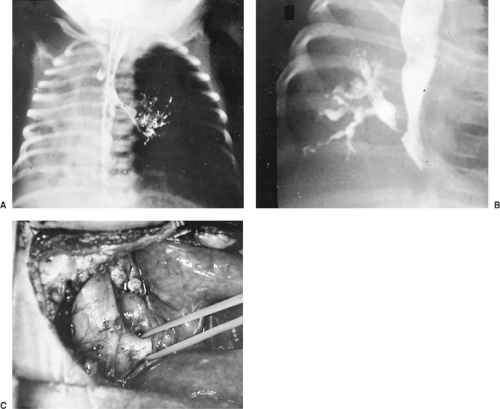 Figure 83-4. A: Atretic right bronchus in a 1-month-old girl who presented with respiratory distress, cough, and fever. B: The esophagogram outlines the right bronchus and lung. C: At surgery, the right esophageal bronchus is identified. A right pneumonectomy was performed. The patient has had significant morbidity in the 4 postoperative years. (From: Raffensperger JG. Congenital malformations of the lung. In Raffensperger JG, ed. Swenson’s Pediatric Surgery, 4th ed. Norwalk, CT: Appleton-Century-Crofts, 1980:700. With permission.) |
Congenital Lobar Emphysema
Congenital lobar emphysema refers to the isolated hyperinflation of a lobe in the absence of extrinsic bronchial obstruction. The left upper lobe is involved most often, followed in incidence by involvement of the right middle lobe. Boland and colleagues6 found hypoplastic cartilage in the bronchus of two of seven patients with lobar emphysema. Lincoln and associates39 described hypoplastic or absent cartilage in 22 of 28 examples reviewed. Scarpelli and Auld65 collected a series showing a 25% incidence of dysplasia of the bronchial cartilage. Stovin68 reported abnormal orientation and distribution of the bronchial cartilage in his series of patients.
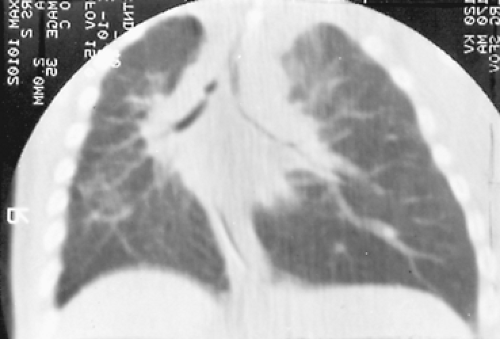 Figure 83-5. Recurrent right-upper-lobe and entire right lung atelectasis in an infant with hyaline membrane disease who required prolonged intubation and ventilation. The right mainstem bronchus was stenotic. Repeated dilations were palliative. This CT scan was obtained to clarify distal anatomy or identify other pathologic change. Note two areas of significant stenosis in the right mainstem bronchus. |
A subset of lobar emphysema is the polyalveolar lobe, first described by Hislop and Reid.28 In an infant with classic congenital lobar emphysema, they found an increase in the number of alveoli in the affected lobe. A subsequent report by Tapper and associates70 reevaluated a group of infants with congenital lobar emphysema and found that 6 of 16 had the abnormal characteristics of the polyalveolar lobe.
Lobar emphysema produces symptoms in infancy. Often, a history of tachypnea, retraction of the chest wall, and wheezing since birth exists. An upper respiratory infection may complicate the condition and precipitate severe respiratory distress. Most children with lobar emphysema present before 6 months of age. Some infants develop symptoms in the first few days of life and require urgent intervention. Physical examination reveals a shift of the trachea and mediastinum to the contralateral hemithorax. Breath sounds are decreased on the affected side, with associated hyperresonance. Radiographs of the chest show hyperaeration of the affected lobe with atelectasis of the adjacent lobes and a mediastinal shift (Fig. 83-6). Careful inspection of vascular markings reduces the risk for misdiagnosis of this lesion as a tension pneumothorax.
In a newborn with severe respiratory distress, a radiograph of the chest is the only preoperative study that is indicated. In an infant with mild to moderate respiratory distress, CT scanning can establish the diagnosis of congenital lobar emphysema by showing the hyperlucent expanded lobe and stretched, attenuated vessels. CT scanning can also exclude extrinsic causes of lobar emphysema, such as vascular anomalies or a mediastinal mass. In selected patients, Markowitz and colleagues46 recommend a radionuclide ventilation/perfusion scan to confirm the absent function of the involved lobe. Stigers66 and Doull16 and their associates recommend bronchoscopy to exclude intraluminal pathology and assess the extent of bronchial collapse during ventilation.
Any infant with moderate to severe respiratory symptoms and a diagnosis of congenital lobar emphysema should be treated with lobectomy. Infants with mild symptoms may be carefully followed, although this management strategy is controversial. At operation, the chest is opened as soon as possible after induction of anesthesia. Positive-pressure ventilation causes further overinflation of the involved lobe and increases the risk for cardiovascular compromise. Gupta and associates22 recommend selective intubation of the contralateral lung with controlled ventilation in these infants. Raghavendran and associates55 recommend continuous caudal epidural anesthesia to avoid the risk of positive-pressure ventilation. The abnormal lobe usually herniates through the thoracotomy incision. The lobe feels like sponge rubber, does not deflate, and bounces back into shape after it is compressed. Its edges are rounded and poorly defined. The remaining lung is atelectatic. Before resection, the mediastinum must be carefully examined for lesions that could have obstructed the bronchus. After lobectomy, the remaining lung expands to fill the chest. The emphysematous lobe characteristically does not deflate even after it is removed from the chest.
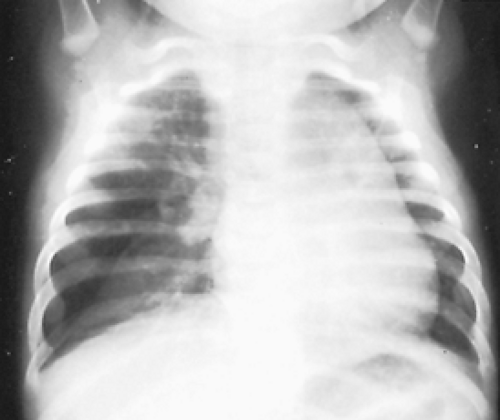 Figure 83-6. Radiograph of a newborn with lobar emphysema involving the right middle lobe. Note the compressed right lower lobe and mediastinal shift. |
Infants with hyaline membrane disease who require prolonged mechanical ventilation may develop acquired lobar emphysema (Fig. 83-7). The right lower lobe is most frequently affected. Suction trauma may cause squamous metaplasia of the bronchial orifice, and repeated barotrauma contributes to the ruptured alveoli and emphysematous changes. Radionuclide scans demonstrate poor perfusion of the affected lobe.
Cooney and colleagues11 recommended lobectomy when the infant could not be weaned from the ventilator because of the acquired emphysematous lobe.
Cooney and colleagues11 recommended lobectomy when the infant could not be weaned from the ventilator because of the acquired emphysematous lobe.
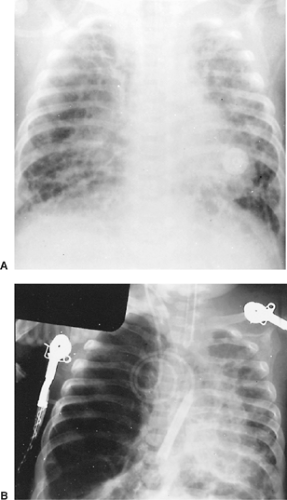 Figure 83-7. A: Radiograph of the chest of a 2-month-old infant with bronchopulmonary dysplasia. B: Within 3 months, lobar emphysema developed in his right lower lobe. Further mediastinal shift and increasing ventilator requirements prompted lobectomy. |
In older infants with a history of respiratory problems, CT may help identify other causes of acquired lobar emphysema, such as extrinsic bronchial compression from enlarged lymph nodes, a bronchogenic cyst, or anomalous blood vessels. In an older child with an acute onset of symptoms, bronchoscopy is indicated to rule out an aspirated foreign body or an endobronchial mass. The bronchoscopy should be planned to immediately precede thoracotomy. Oxygen, antibiotics, and humidity are administered prophylactically.
Pulmonary Dysplasia
Pulmonary Agenesis and Aplasia
Unilateral pulmonary agenesis results from lack of development of a single lung bud. Lung parenchyma and pulmonary vessels are lacking. Sbokos and McMillan64 reported a 50% incidence of associated cardiac disease, especially when the agenesis was on the right side. Say and associates63 noted that pulmonary agenesis may be associated with a chromosomal abnormality (46,XX,2p+).
A newborn with unilateral pulmonary agenesis may be asymptomatic or present with tachypnea, dyspnea, and cyanosis. Older infants or children may present with wheezing that suggests asthma or bronchitis. On physical examination, the trachea and mediastinal structures are shifted to the involved side. The overall shape of the chest is normal. Signs of airway obstruction and poor bronchial drainage may be recognizable. A radiograph of the chest reveals absence of lung markings and mediastinal shift to the ipsilateral side (Fig. 83-8). The differential diagnosis includes total lung atelectasis, total lung sequestration, or lung with an esophageal bronchus. The normal position of the diaphragm and normal intercostal spaces preclude atelectasis. An esophagogram and CT scan exclude the other possibilities. Echocardiography helps in diagnosing the associated cardiac lesions. No particular treatment exists for pulmonary agenesis, but correction of the cardiac anomaly may relieve some of the symptoms. Massumi and associates48 reported that 30% of infants with agenesis die in the first year of life and 50% die within the first 5 years. The mortality rate was higher with right-sided agenesis. Maltz and Nadas43 reviewed the world literature and found 164 cases of pulmonary agenesis. Of the 36 patients reported by 1954, a total of 24 were alive in 1968.
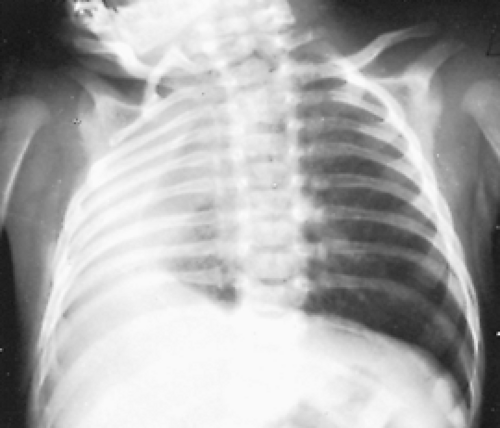 Figure 83-8. Radiograph of the chest of a newborn with respiratory distress and agenesis of the right lung. There is a marked mediastinal shift to the ipsilateral side. |
Bilateral pulmonary agenesis is incompatible with life, although Claireaux and Ferreira9 reported one infant who survived for 15 minutes. The trachea ends blindly or into primitive lung tissue. The incidence of associated anomalies is high.
Pulmonary aplasia is similar to pulmonary agenesis in that pulmonary parenchyma and vessels are absent. The blind bronchial stump may serve as a source of repeated infection in the normal contralateral lung. The stump may be identified by bronchoscopy or CT scan. Once recognized, the chronically infected stump should be resected (Fig. 83-9).
Primary Pulmonary Hypoplasia
Pulmonary hypoplasia can be identified pathologically by the radial alveolar count and the ratio of lung weight to body weight. Pulmonary hypoplasia is primary if no obvious cause for it can be found. Swischuk and associates69 reported that four of eight infants with primary pulmonary hypoplasia had mean radial alveolar counts lower than normal and thickening of the pulmonary arteriolar wall. The hypertrophy of the muscular layer of the pulmonary arteriole develops in response to
fetal stress. This hypertrophy is frequently found in infants with pulmonary hypoplasia and other causes of pulmonary hypertension. Haworth25 suggested that the normal postnatal regression of the pulmonary arteriolar muscle does not occur.
fetal stress. This hypertrophy is frequently found in infants with pulmonary hypoplasia and other causes of pulmonary hypertension. Haworth25 suggested that the normal postnatal regression of the pulmonary arteriolar muscle does not occur.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


