Chapter 59 Congenital Heart Disease
History and Other Considerations
The era of surgical treatment for congenital cardiac anomalies was initiated in November 1944, when Dr. Alfred Blalock and associates Vivien Thomas and Dr. Helen Taussig combined their unique talents and vision to treat a young child dying of cyanotic congenital heart disease (CHD).1 This palliative operation involved the surgical creation of a systemic to pulmonary artery connection in the patient suffering from inadequate pulmonary blood flow. The procedure has since been recalled as miraculous and has carried the eponym of the Blalock-Taussig shunt (BT shunt) during the ensuing years, now more than 60 years later. The striking success of this simple concept and the reproducible nature of the operation in children suffering from otherwise fatal cardiac conditions has emboldened subsequent surgical innovators to venture inside the congenitally malformed heart. At first, a parent was asked to serve as a biologic oxygenator using the technique of controlled cross-circulation; soon thereafter, the mechanical, extracorporeal, heart-lung bypass pump was developed.2,3 With the aid of this ability to support the patient’s circulation during intracardiac exploration, surgeons have sequentially attacked almost every described congenital cardiac anomaly. The prospect of meaningful survival for patients born with otherwise devastating congenital cardiac lesions is now expected in most, if not all, cases.
As a result of this success story, there is now a large and growing population of adults with repaired or unrepaired CHD; estimates in the United States for 2005 placed the number of adult patients surviving with repaired or palliated congenital cardiac lesions at more than 1 million persons.4 This reality has been associated with new challenges in the ongoing medical maintenance of such patients, with particular focus on the care of patients with congenital cardiac lesions presenting for surgery for noncardiac illnesses. The evolving subspecialty of adult CHD points to the unique needs of this population of patients.
Pathways For Practicing Congenital Heart Surgery
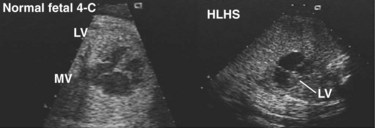
Before embarking on a review of the field, it is worthwhile to describe the setting in which patients with CHD seek and receive care in today’s medical environment. With the development of sophisticated methods of fetal ultrasound, a large percentage of children requiring surgery for CHD are diagnosed during gestation (Fig. 59-1). Although not yet confirmed as affecting overall survival rates, a fetal diagnosis of complex CHD is of inordinate help to parents and the medical management team. This is particularly important in the setting of lesions dependent on persistent patency of the ductus arteriosus for postnatal survival. In these individuals, survival after delivery is predicated on the maintenance of ductal patency through the IV infusion of prostaglandin E1 (PGE1) initiated in the delivery suite, often through an umbilical vein catheter.
Historically, pediatric cardiologists have provided the medical management of patients born with CHD. Pediatric cardiology is also evolving. With advances in catheter-based technology, lesions previously treated with surgery are now being addressed by interventional pediatric cardiologists. Examples include device closure of atrial and ventricular septal defects (VSDs), occlusion of patent ductus arteriosus (PDA), and dilation and stenting of stenotic vessels in the systemic and pulmonary circulation. For a more in-depth recent review of this specialty, see the excellent technical text by Mullins.5
On the other hand, pediatric cardiologists are not educated in adult medicine and cardiology; many feel uncomfortable providing consultation on adult patients with CHD. As noted, the subspecialty of adult CHD is still in a state of development but, at present, there are few physicans who have been educated specifically to care for these patients. This underscores the necessity of the practicing general surgeon to become familiar with the specific issues of concern for patients with CHD to ascertain that the patient’s unique anatomic and physiologic issues have been evaluated properly. Adult patients with CHD who present for care in a center without a designated qualified specialist must be evaluated by a pediatric cardiologist in coordination with an adult cardiologist. Of equal importance, the anesthesiologists and intensivists caring for such a patient must have a working understanding of the complexities and nuances of the patient’s cardiac condition.6 The anesthetic management of patients with CHD undergoing general surgical procedures is complicated and can become disastrous if managed improperly.
Anatomy, Terminology, and Diagnosis
Anatomy and Terminology
One of the most intimidating aspects for the student of CHD is developing a level of comfort with the terminology used for describing specific lesions. To begin, a thorough and sound understanding of normal cardiac anatomy is mandatory. There are several excellent texts on this subject; in particular, the one edited by Wilcox and coworkers7 is especially concise and clear. One difficulty that challenges proper understanding of anatomy is the frequent use of abbreviations and eponyms for various congenital lesions—for example, congenitally corrected transposition of the great arteries (ccTGA), ventricular inversion, and L-transposition all describe the same heart, but none provides a complete anatomic description. Unless otherwise clear to all involved in the care of these complicated patients, the anatomic description needs to be segmental and complete to avoid mistakes and misinterpretations of structure.
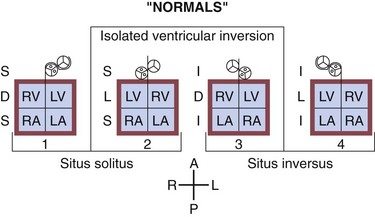
There are two widely accepted and applied schools of cardiac morphologic description. The Van Praagh nomenclature uses abbreviations to describe the relationship of the atria, ventricular looping, and position of the aorta sequentially. The first letter describes the situs of the atrial chambers (and usually the abdominal organs): S for situs solitus (normal), I for situs inversus (reversed), or A for situs ambiguous (indeterminate). The second letter describes the relationship of the embryologic looping of the ventricles; D for dextro looping or right-handed topology (normal) or L (levo) for left-handed topology. The third and last letter describes the relationship of the aortic valve to the pulmonary valve, D for right-sided and L for left-sided (Fig. 59-2).
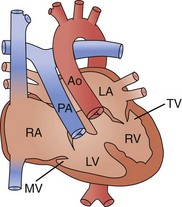
The Anderson nomenclature is more wordy and longer, but is perhaps simpler to understand. The descriptions are again of the sequential relationship of the structures. Starting with the atria, the connections and relationships are sequentially described. Thus, the atrial sidedness is described, followed by the sequence of connections to the ventricles and then great vessels. For example, atrial situs solitus (normal) with atrioventricular discordance (reversed) and ventriculoarterial discordance (reversed) describes the heart mentioned earlier as corrected transposition, or S,L,L by the Van Praagh classification (Fig. 59-3).
Diagnosis
Diagnostic Tests
Plain Radiography
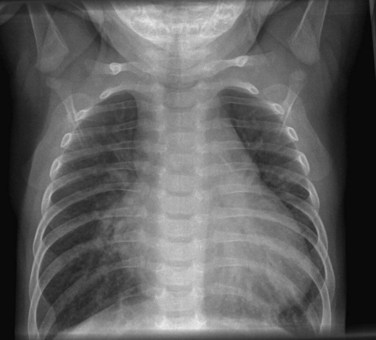
Standard chest radiography with anteroposterior and lateral views is still an essential component of the assessment of a patient with CHD. Standard elements to be examined include a skeletal survey, assessment of the diaphragms and hepatic shadow, and location of the gastric bubble. The lung fields are assessed for pulmonary plethora (arterial or venous), air space disease, and presence of effusions. The cardiac silhouette may reveal much important information, such as a cardiothoracic ratio indicative of cardiomegaly or pericardial effusion, presence of atrial enlargement, presence or absence of the pulmonary artery shadow, and arch sidedness (Fig. 59-4).
Cardiac Catheterization
Usually, however, diagnostic cardiac catheterization is performed to obtain precise hemodynamic information needed to make an informed assessment of the consequences of the patient’s cardiac lesions. Using oximetric measurements, pressure data, and thermodilution cardiac output determination, accurate assessment of the patient’s hemodynamic profile is obtained. Measured or derived data include central venous pressure, atrial pressure, ventricular pressures (including end-diastolic pressure), shunt fraction (in the case of atrial or VSDs), pulmonary artery pressures, pulmonary capillary wedge pressure, systemic arterial pressure, and segmental oximetry of cardiac structures, including systemic and pulmonary venous return (Fig. 59-5). Thus, critical information is obtained about the presence and degree of shunting, systemic and pulmonary vascular resistance, and cardiopulmonary function. In certain clinical settings, these data are mandatory to a successful clinical management strategy. This may be particularly true for the adult patient with congenital cardiac disease requiring noncardiac surgery.
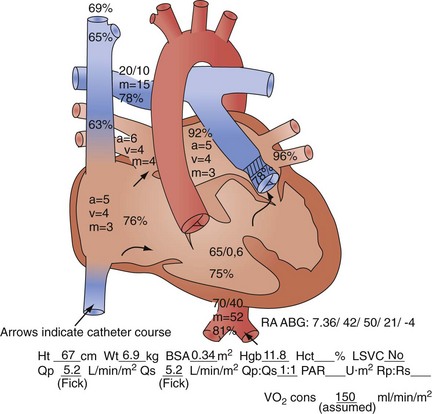
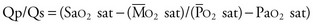
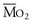 sat is mixed venous oxygen saturation,
sat is mixed venous oxygen saturation, 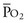 sat is pulmonary venous oxygen saturation, and Pa
sat is pulmonary venous oxygen saturation, and PaThus, in a patient with 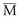 sat of 60%,
sat of 60%, 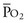 sat of 100%, Sao2 sat of 100%, and PaO2 sat of 80%,
sat of 100%, Sao2 sat of 100%, and PaO2 sat of 80%,
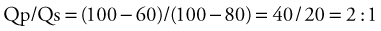
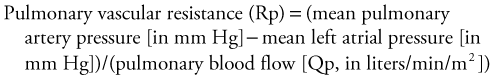
Perioperative Care
Anesthesia Pitfalls
Providing physiologic anesthetic management can be challenging in patients with congenital cardiac disease, especially in situations such as chronic single-ventricle palliation, unrepaired CHD, chronic cyanosis, and residual intracardiac pathology. It is important to note that standard anesthetic management paradigms may be completely inappropriate and potentially disastrous in the setting of complex CHD. A thorough understanding of the patient’s anatomy is mandatory, along with knowledge of the potential for unexpected response to anesthetic agents and ventilator settings. The field of pediatric and congenital cardiac anesthesia has evolved relative to this specific clinical need; the recent text by Andropoulos and colleagues is an excellent resource.8
Ventilator management in the perioperative setting of CHD requires special understanding. In settings of large potential left-to-right shunts (e.g., unrepaired VSDs), hyperventilation and hyperoxygenation will promote excessive pulmonary blood flow and potentially diminish systemic cardiac output. Positive pressure ventilation, particularly positive end-expiratory pressure (PEEP), will negatively influence hemodynamics in many patients, especially in palliated single ventricle patients after the Fontan procedure. Early extubation in this population can be done to limit the deleterious effects of PEEP on the Fontan circulation. Early data have shown that this improves outcomes for these patients and reduces overall hospital costs.9 Finally, pharmacologic manipulation of systemic and pulmonary vascular resistance and cardiac performance is an important adjunct in the perioperative management of patients with CHD. In general, a low-dose infusion of epinephrine (0.05 µg/kg/min) with the addition of a phosphodiesterase inhibitor is an effective pharmacologic cocktail to promote cardiac inotropic state, lower systemic and pulmonary vascular resistance, and limit tachycardia. Other agents frequently used include dopamine, vasopressin, sodium nitroprusside, and nitroglycerin. Appropriate perioperative analgesia and sedation are also important aspects of the patient’s management.10
Neurologic Outcomes
With expectations of almost 100% survival following surgery for CHD, emphasis has been placed on the long-term neurologic outcomes and quality of life of these patients. The potential for neurologic insult in children following CHD arises from the nature of their disease (e.g., cyanotic defects, low cardiac output state, genetic syndromes, effects of cardiopulmonary bypass, circulatory arrest). There is also evidence to suggest that perhaps patients with CHD are genetically predisposed to neurologic insult. Recently, gestational age has been found to be an important factor to consider in the optimization of neurologic outcomes.11
Lesion Overview
Defects Associated With Increased Pulmonary Blood Flow
Persistent Arterial Duct (Patent Ductus Arteriosus)
The gold standard of therapy for closure of PDA is surgery, usually accomplished through a left thoracotomy using ductal division, ligation, or clipping (Fig. 59-6). This needs to be a low-risk procedure associated with minimal potential for persistence. Nonetheless, the invasive nature of this proven method has led to the development of alternative strategies for ductal occlusion. From a surgical perspective, many PDAs are amenable to thoracoscopic clipping through very small port incisions; robot-assisted PDA occlusion has been performed in many patients, with good results.12 At present, however, most PDAs are occluded in the cardiac catheterization laboratory using occlusive devices. Even the repair of large defects in small babies has been successfully addressed. The long-term effects of the devices remaining in the vascular tree are as of yet not fully understood; however, successful device closure appears to be an extremely effective and durable therapy.13
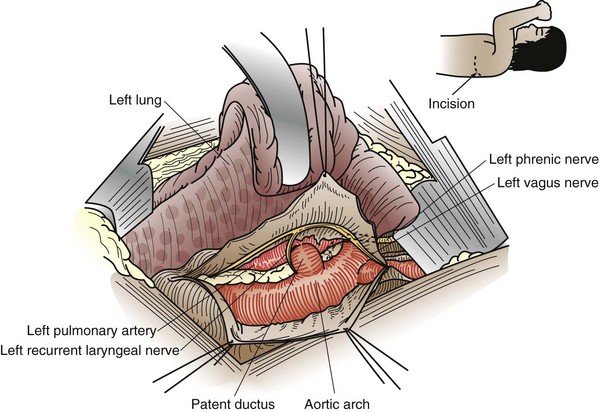
FIGURE 59-6 Anatomic relationships of a patent ductus arteriosus, exposed from a left thoracotomy.
(From Castaneda AR, Jones RA, Mayer JE Jr, Hanley FL: Patent ductus arteriosus. In Castaneda AR, Jones RA, Mayer JE Jr, Hanley FL: Cardiac surgery of the neonate and infant, Philadelphia, 1994, WB Saunders.)
Aortopulmonary Septal Defect (Aortopulmonary Window)
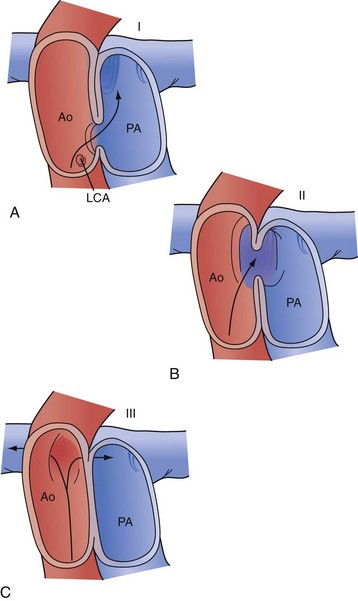
An aortopulmonary septal defect is a communication between the ascending aorta and, usually, the main pulmonary artery. This is a rare defect; it relates to the common embryologic origin of the arterial trunk and failure of complete separation into the aorta and pulmonary artery. Defects are classified by their location: type I is proximal, just above the aortic sinuses; type II is more distal on the ascending aorta and often involves the origin of the right pulmonary artery; and type III is more distal and associated with a separate origin of the right pulmonary artery from the aorta (Fig. 59-7). An aortopulmonary septal defect may occur in isolation or in association with other conditions, including interrupted aortic arch (IAA) and anomalous origin of a coronary artery. Defects are typically large and responsible for a large left-to-right shunt with systemic pulmonary artery pressures. Children with this problem typically present with CHF, failure to thrive, and frequent respiratory infections. Diagnosis may be made by echocardiography, MRI, or catheterization.
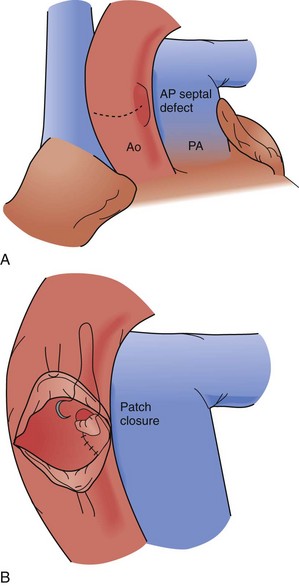
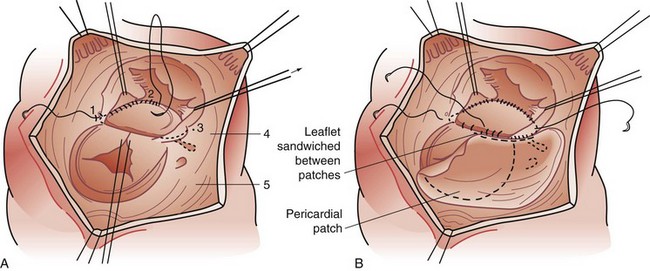
All aortopulmonary septal defects are surgically closed; this lesion is not amenable to catheter-based closure and such an attempt is hazardous. A small defect may be ligated through a thoracotomy or median sternotomy approach, but this method is not recommended because of significant risk for rupture or incomplete closure. Surgical closure is accomplished with cardiopulmonary bypass support. Options for closure include complete division and separate patch repairs of the great vessel defects or a sandwich type of closure, using a patch to construct a common intervening wall; both methods are effective (Fig. 59-8).
Atrial Septal Defect
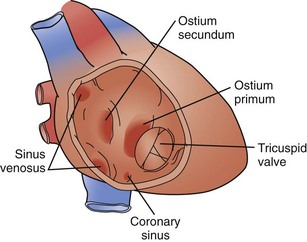
An isolated atrial septal defect (ASD) is one of the most common congenital cardiac lesions. The most frequently encountered ASD relates to a defect in the interatrial wall, as defined by the fossa ovalis. The defect develops as the result of incomplete closure of the embryologic patent foramen ovale; thus, the defect is a result of incomplete closure of the septum primum. Although the terminology can be confusing, these defects are typically termed secundum atrial septal defects. They present in a wide variety of configurations, ranging from single small defects to multiple fenestrations to complete absence of the septum primum. The confines of the defect may extend from the IVC orifice up to the superior atrial wall adjacent to the aortic root (Fig. 59-9).
Most centers recommend ASD closure before school age. The standard therapy for ASDs since the late 1950s has been surgical closure using cardiopulmonary bypass support. The defect is closed using direct suture closure, autologous pericardium, or prosthetic patch material (Fig. 59-10). This is an effective method, with a low associated perioperative risk, including the virtual absence of residual or recurrent defects, as noted in one study.14 Minimally invasive techniques for ASD closure have also gained popularity.
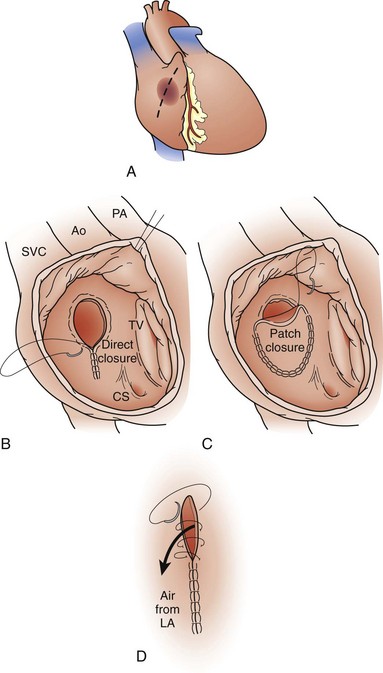
The potential for closing defects using nonsurgical methods has led to the development of catheter-based therapies, which are now being widely applied to large numbers of patients worldwide for the treatment of ASD. The most commonly used device is the Amplatzer septal occluder (St. Jude Medical, St. Paul, Minn) device, of nitinol metal mesh, which is placed percutaneously and delivered with echocardiographic and fluoroscopic guidance. Early reports have indicated an acceptable procedure-related complication rate and successful closure rate.15 It is clear, however, that the long-term effects of having such a device in mobile cardiac structures are not fully understood. Several recent reports have documented an alarming incidence of device erosion through the atrial wall and into the adjacent ascending aorta, as well as disruption of the conduction system.16,17 A recent case of late severe endocarditis involving a previously placed Amplatzer ASD device has highlighted the need for ongoing observation of the long-term consequences of placing large prosthetic devices into the circulation.18
Sinus venosus atrial septal defects occur as the result of embryologic malalignment between the superior vena cava (SVC) or IVC. These defects are not associated with the ovale fossa and are frequently associated with partial anomalous pulmonary venous return. A superior sinus venosus ASD occurs high in the atrium, near the orifice of the SVC. This lesion is frequently associated with anomalous drainage of a portion of the right lung to the SVC. An inferior sinus venosus ASD is located low in the atrium, often extending into the IVC orifice. This lesion is typically associated with anomalous pulmonary venous drainage of the entire right lung to the IVC (potentially intrahepatic); pulmonary sequestration and an abnormal systemic artery perfusing the right lower lobe, with origin from the abdominal aorta, may also be present. In patients with total anomalous pulmonary venous return (TAPVR) to the IVC, the anomalous pulmonary vein may be readily obvious on a plain chest radiograph and has been described as appearing like a saber (scimitar syndrome), first described by Sabiston and Neill.19
Surgery for sinus venosus ASDs is recommended for the same pathophysiologic reasons as secundum ASDs. The repair is not amenable to catheter techniques, and surgery is more complicated than for an isolated secundum ASD. Superior sinus venosus defects with partial anomalous pulmonary venous return to the SVC may be treated with an intracardiac patch baffle; however, in the setting of high drainage of the anomalous pulmonary veins, an SVC translocation operation (Warden procedure) may be necessary.20 Surgery for an inferior sinus venosus ASD with a scimitar vein can be more complicated, potentially involving the need for a patch baffle within the intrahepatic IVC, which may require periods of hypothermic circulatory arrest.
Ventricular Septal Defect
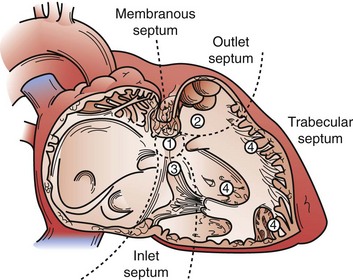
Subarterial (Supracristal or Outlet) Ventricular Septal Defect
Subarterial VSDs occur in association with the annulus of the aortic valve, pulmonary valve, or both. The defects are almost always associated with significant prolapse of the adjacent aortic valve cusp, usually the right coronary cusp, which may lead to significant cusp distortion, aortic valve insufficiency, and even cusp perforation. The only mechanism for spontaneous closure of these defects relates to the cusp prolapse and valve distortion and is generally not complete or a favorable arrangement. All these defects are surgically closed because of the ongoing risk for aortic valve injury (Fig. 59-11).
Although catheter-based therapies for some VSDs have been developed, particularly muscular defects, this mode of therapy is still not widely applicable to most VSDs.21 The complex relationship of many defects, including close association with the aortic valve and cardiac conduction tissue, makes the existing technology less than ideal. At present, surgery remains the primary mode of therapy for VSD closure. Defects are approached with the aid of cardiopulmonary bypass support and may be closed with various materials, including autologous pericardium (our preference), Dacron, polytetrafluoroethylene (PTFE), and homograft material. Surgical closure of VSDs is a low-risk procedure with a high expectation of complete closure.22 Challenging anatomic situations such as Swiss cheese septum or multiple apical muscular VSDs may be initially palliated by limiting pulmonary blood flow with a pulmonary artery band and deferring corrective surgery to later in life.
Atrioventricular Septal Defect (Atrioventricular Canal Defect)
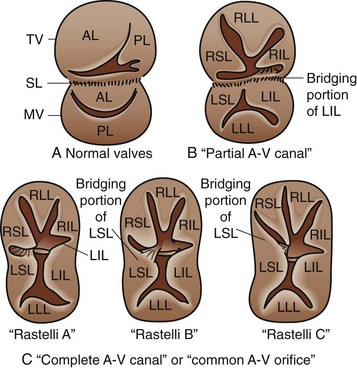
Atrioventricular septal defects (AVSDs) are a complex constellation of cardiac lesions involving deficiency of the atrial septum, ventricular septum, and atrioventricular valves. This lesion results from an embryologic maldevelopment involving the endocardial cushions; thus, the term endocardial cushion defect is often applied. AVSDs may be partial, involving no ventricular level component, intermediate or transitional, involving a small restrictive VSD, or complete, involving a large nonrestrictive VSD. The atrioventricular valve tissue is always abnormal in AVSD, although there is great individual variability in terms of the severity of the valvular malformation and thereby valve function. Complete AVSDs are frequently seen in patients with trisomy 21, but do occur in patients with normal chromosomes. The morphology of the septal defects in this condition is different than that previously discussed. The ASD in this defect is termed a primum ASD and is distinctly separate from the ovale fossa. There is displacement of the atrioventricular node and bundle of His to the inferior aspect of the primum defect and atrioventricular junction, a feature of critical importance during surgical repair. Patients with AVSD have an inlet VSD, which may extend into the subaortic region and have a component of septal malalignment. The chordal support of the atrioventricular valves has a variable relationship to the interventricular septum. The relationship of the chordal support and superior bridging component of the left atrioventricular (AV) valve have been used to classify complete AVSD, as described by Rastelli and associates23: type A with superior leaflet and chordal support committed to the left side of the ventricular septum; type B with straddling, shared chordal support; and type C with a floating left superior leaflet component and chordal support on the right side of the ventricular septum (Fig. 59-12).
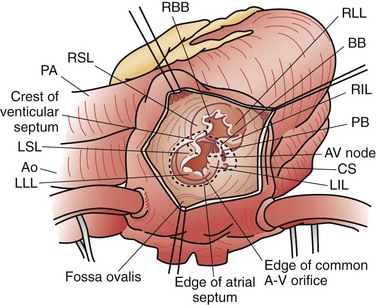
Surgery is the primary mode of therapy for patients with AVSD. Operative goals include complete closure of ASDs and VSDs and effective use of available AV valve tissue to achieve valve competence. As noted, the inferiorly displaced conduction tissue must be protected to avoid the complication of surgically induced AV block (Fig. 59-13). Patients are approached with the aid of cardiopulmonary bypass support. The atrial and ventricular septal components are closed with a common patch (single-patch method) or separate patches (two-patch technique). We believe the two-patch method to be superior in preserving AV valve tissue (Fig. 59-14).24 The critical component of the repair lies in the valve repair; typically, after suspending the valve tissue to the reconstructed septum, the line of coaptation between the superior and inferior leaflet components (cleft) is closed. However, care must be exercised to avoid valvular stenosis.
Persistent Arterial Trunk (Truncus Arteriosus)
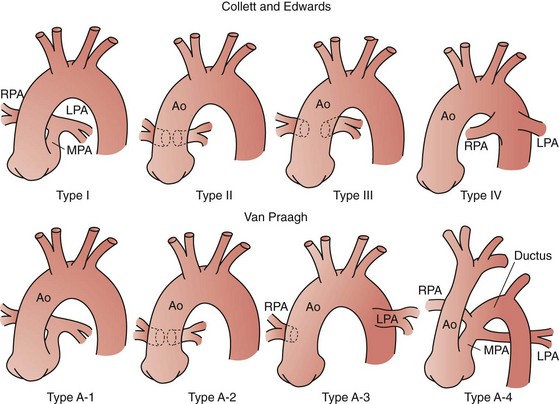
Truncus arteriosus or persistent arterial trunk results from failure of separation of the embryonic arterial trunk and semilunar valves. It is almost always associated with a large nonrestrictive VSD, is typically perimembranous, and is associated with varying degrees of truncal override of the interventricular septum, including 100% association of the trunk with the right ventricle. The condition is classified by the relationship of the origins of the pulmonary arteries: in type I truncus arteriosus, there is a demonstrable common main pulmonary artery with subsequent origins of the branch pulmonary arteries; in type II truncus arteriosus, the branch pulmonary arteries arise closely, but separately, from the trunk; and in type III arteriosus, the branch pulmonary arteries are widely separated in origin on the ascending aorta (Fig. 59-15).
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


