CHAPTER 33 Congenital Diaphragmatic Hernia
HISTORY
The first known written description of a diaphragmatic herniation was made by Ambroise Parè in 1579.1 However, the two cases described by Parè were caused by trauma. In his 1679 Sepulchretum, Teophile Bonet attributed the first description of a congenital case to Lazare Rivere, at the beginning of the 17th century, even though it was from the autopsy of an adult.2 The first report of a CDH in a newborn was made by George Macaulay, in 1754, also as an autopsy finding, in an infant who died from respiratory failure a little over 1 hour after birth.3
In 1761, Giambattista Morgagni, then a pupil of Valsalva, wrote a review of diaphragmatic herniations and credited Stehenius with the first observation that CDH was associated with pulmonary hypoplasia.4 In the review, Morgagni described the first case of a parasternal hernia, henceforth termed Morgagni hernia, in an elderly man. In 1848, Vincent Alexander Bochdalek reported two cases, and he described the location of the diaphragmatic defect as being in the posterolateral aspect of the muscle—hence the origins of the terms Bochdalek hernia and Bochdalek foramen.5 Although widely employed, these terms are actually improper, as the mechanisms and exact location proposed by Bochdalek—namely, rupture of the lumbocostal triangle—are inaccurate.6–8
The link between CDH and deviations of the embryonic development of the pleuroperitoneal membrane only started to be established from the studies by Broman, in 1902 and 1905.9,10 Nevertheless, although this anatomic location for the diaphragmatic defect is universally accepted, it is not yet known where the primary disorder that leads to CDH takes place.
The first successful repair of a CDH was performed by Aue in a 9-year-old boy in 1901, but it ws published only in 1920.11 The first publication of a successful CDH repair was by Heidenhain, in 1905, and he reported a procedure, also in a 9-year-old boy, that took place in 1902.12 In 1940, the first survival of a neonate, who underwent CDH repair on the second day of life, was reported by Ladd and Gross.13 Gross was also the first to report on a good outcome after repair before the first 24 hours of life, in 1946.14
In 1977, German and coworkers reported the first child with CDH to survive after being placed on extracorporeal membrane oxygenation (ECMO).15 Since the mid 1980s, ECMO has played a major role in maximizing the survival rates of neonates with CDH.16–18
In 1989, Harrison and associates presented the first series of patients with CDH treated by open prenatal repair of the diaphragmatic defect in humans, leading to live births but no midterm survival.19 Survival after this procedure was reported by this same group in 1992, but only in 28.6% of the operated fetuses.20
In the early 1990s, Wung and colleagues introduced the concept of avoiding hyperventilation and minimizing barotrauma.21,22 This led to marked reduction in iatrogenic insult to the lungs, which was very common with the old, aggressive ventilation strategies, and the consequent improvement in survival has been significant.16,23–25
The first successful lung transplantation for the treatment of CDH was performed in a newborn in 1992, by the groups of Shochat and Starnes.26,27 Also in 1992, Wilson first showed, in an ovine model of CDH, that fetal lung growth could be significantly accelerated after occlusion of the fetal trachea, leading to reversal of the pulmonary hypoplasia associated with experimental CDH.28 In 1994, Harrison’s group applied this maneuver successfully in a human fetus with CDH.29 Nonetheless, the results of fetal tracheal occlusion, whether performed as an open or a video-fetoscopic procedure, remain worse than those of postnatal care, at least at the referral centers in North America.30–32 Still, fetal tracheal occlusion continues to be offered systematically at a few European centers, and (anecdotally) elsewhere, although its current indications, if any, remain to be clearly defined.33,34
In 1994, the Congenital Diaphragmatic Hernia Registry was created as a meta-institutional organization dedicated to the exchange and analysis of data related to this disease, as well as to the design and implementation of multicenter prospective trials. This initiative, molded after the pediatric oncology groups first established in the 1980s, has had a major beneficial impact on CDH survival and on management guidelines.35
In 1995, we demonstrated experimentally that lung growth could be accelerated after birth, through continuous intrapulmonary distention with a perfluorocarbon.36,37 The first series of patients that received this treatment, under ECMO support, was presented in 2000.38 A multicenter prospective trial of this principle has proven difficult to pursue because of regulatory and logistical hurdles, yet it remains an anticipated perspective. Also in 2000, the use of a tissue-engineered construct for the repair of the diaphragmatic defect was first proposed in an animal model.39 Further experimental developments have validated engineered diaphragmatic repair, and a clinical trial is expected in the foreseeable future, pending regulatory clearance.40–43
EPIDEMIOLOGY
The prevalence of CDH has been reported as being between 1:1200 and 1:12,000 births.44–56 The main reason for such disparity is probably the so-called hidden mortality of CDH, which means that many babies die before reaching a referral center and thus are not included in the statistics.57 The better-controlled studies show CDH as occurring in 1:2107 to 1:3163 births.44,47,49,50,52,54 One of the largest series, by Torfs and colleagues, involving over 718,000 births and stillbirths in California, revealed that CDH occurred in 1:3163 births and in 1:3340 live births.54 Among the major congenital anomalies, CDH is one of the most common, accounting for approximately 8% of the cases.58
There are no clear racial differences in the prevalence of CDH.52,54 The study by Torfs and coworkers showed a higher prevalence in rural rather than urban areas, but this has not yet been confirmed by other series.54 The prevalence of prematurity and weight deviations in neonates with CDH is no different from that in the general population.54 Although isolated CDH is a little more common in boys than in girls (1.5:1), sex distribution is equal in CDH associated with other congenital anomalies and in the whole CDH population.54,59
There is limited controversy as to the risk of CDH recurrence in families. In the vast majority of cases, CDH is sporadic, with no genetic component described and with the risk for further offspring being practically equal to that in the general population.44,47 At the same time, many families with more than one child with CDH, usually siblings, have been identified.49,54,58,60–66 Less frequently, relatives other than siblings present with this anomaly.61,67,68 The epidemiologic profile of familial cases of CDH differs little from that of the total of cases: it is slightly more common in males (approximately 2:1),60,63,67,69 and the data are conflicting as to other variables.62,68 The occurrence of familial CDH follows a pattern suggestive of multifactorial inheritance as the most likely mode of transmission, with recurrence estimated between 1.3% and 2%.54,58,62,65 The possibility that a genomic imprinting phenomenon takes place has also been proposed.66 A recent extensive review of a single-hospital-based malformation surveillance program (the largest to date of consecutively collected cases of CDH) showed low precurrence among siblings.70
EMBRYOLOGY OF THE DIAPHRAGM
The development of the diaphragm from the mesoderm is complex and not yet fully understood. It results from the fusion of four embryonic components: two odd ones—the transverse septum and the mediastinum (also known as the dorsal mesentery of the esophagus)—and two paired structures—both sides of the body wall musculature and both pleuroperitoneal membranes (Fig. 33-1).9
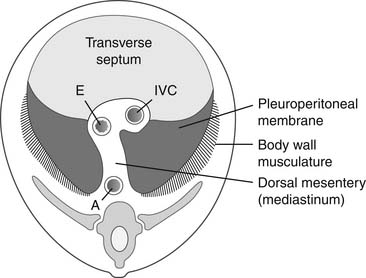
Figure 33–1 The four embryonic components of the diaphragm. A, aorta; E, esophagus; IVC, inferior vena cava.
The diaphragm begins to develop during the 3rd and 4th weeks of gestation, with the appearance of the first component of the diaphragm, the transverse septum (see Fig. 33-1). At this point, the transverse septum is an incomplete mesenchymal divider, related cranially to the pericardial cavity and caudally to the midgut. Dorsally, the transverse septum blends with the mediastinum. On each side of the mediastinum there are the pleural canals, which connect the pericardial and peritoneal cavities. The subsequent development of the diaphragm depends on the closure of these dorsal pleural canals, which will give rise to the pleural cavities.
During the 4th week, the pulmonary buds, which are developing inside the mediastinum, begin to protrude into the pleural canals. At this stage, the pleural canals are very small and the pericardial cavity is very large. Crests formed on both extremities of the pleural canals will separate the future pleural cavities from the pericardial cavity, cranially, and from the peritoneal cavity, caudally. The cranial crest will give rise to the pleuropericardial membrane and the caudal crest will form the pleuroperitoneal membrane (see Fig. 33-1).
The enlargement of what are now the pleural cavities leads to a progressive narrowing of the opening between them and the pericardial cavity, as well as to the development of the pleuropericardial membrane. In like manner, there is progressive narrowing of the pleuroperitoneal canals and the development of the pleuroperitoneal membranes.6,7 The definitive closure of the pleuroperitoneal canals, now small, will occur during the 8th week of gestation.6
After closure of the pleuroperitoneal canals, the pleural cavities continue to expand, in parallel to lung growth. Cranially, they spread out beyond the limits of the pericardial space. Caudally, they extend into the body wall.6 During this process, which takes place from the 9th to the 12th week, the mesoderm in the posterior thoracic wall appears to be carved out by the caudal borders of the expanding pleural cavities, so that its inner portion will become part of the diaphragm. At the same time, a similar process happens at another, more lateral and anterior portion of both the thoracic and the abdominal walls. As a consequence, part of the diaphragmatic muscle originates from the musculature of the thoracic and the abdominal walls.6,71
Despite the almost universal acceptance of this explanation for the development of the diaphragmatic muscle, there is some controversy related to the role of the phrenic nerve in that process. In accordance with the principle that muscles in general retain their original segmental innervation, certain authors believe that myoblasts derived from the caudal portion of the infrahyoid mesoderm migrate, together with the phrenic nerve, from the third and fourth cervical somites, toward the diaphragm, so that this would be the origin of the diaphragmatic muscle.72,73 However, the phenomenon of myoblasts migrating together with the phrenic nerve has not yet been proven; it is but a partially accepted theory. The fact that the tendinous center of the diaphragm is a fibrous structure completely devoid of muscle fibers speaks against this theory.7 In any event, even if myoblasts from the superior cervical myotomes do follow the phrenic nerve, at least part of the diaphragmatic innervation should be later transferred to muscular portions derived from the thoracic and abdominal walls.
Deviations from the normal development of each one of the various components of the diaphragm bring about different variants of diaphragmatic anomalies. Table 33-1 shows the diverse embryonic origins of the diaphragm and their relationship to different diaphragmatic defects.
PATHOLOGY
Etiology
The etiology of CDH is unknown. In a few rare syndromes in which a diaphragmatic defect is present, there is a well-defined genetic cause, such as in trisomies of chromosomes 13 and 18. On the other hand, a recent single-institution review showed that 17% of all cases of CDH (both isolated and in association with other congenital anomalies) had a recognizable genetic etiology.70 In addition, there was no concordance for CDH among five monozygotic twin pairs. These findings, in conjunction with previous reports of de novo dominant mutations in CDH patients, suggest that new mutations may be an important mechanism in the etiology of this disease. The twin data also point to the possibility that epigenetic abnormalities contribute to its development. Different strategies to reveal eventual CDH-critical chromosome loci and candidate genes in humans are now being actively pursued.74
Experimental CDH can be produced in diverse animal species through different interventions, other than surgical creation of the defect, including exposure to diet deficient in either vitamin A,75,76 zinc,77 or cadmium78; administration of either thalidomie,79 antirat rabbit serum,80 2,4-dichlorophenyl-p-nitrophenyl ether (nitrofen, a herbicide),81–83 or polybromate biphenyls84,85; and genetic manipulations, such as FOG-2, COUP-TFII, and GATA-4 mutations.86–88 However, a conclusive relationship between these experimental models and clinical or epidemiologic data in humans has not been shown.
Pathogenesis
The pathogenesis of CDH is also unknown. Normally, before the return of the bowel from the umbilical cord to the abdominal cavity, which occurs during the 10th week of gestation, it is necessary for the pleuroperitoneal canal and the lumbocostal triangle to firmly close, which happens between the 8th and 10th weeks. If such closure does not take place, the bowel will pass through the pleuroperitoneal canal, sometimes also through the lumbocostal triangle, and will invade the chest, resulting in a “herniation” without a hernia sac. If there is only a membranous closure, the same phenomenon will happen, albeit with a hernia sac that may, or may not, rupture sometime later. In any case, although it is well established that the diaphragmatic defect is at the level the pleuroperitoneal canal,6,7,9,10 the sequence of events that culminates in disturbances of its closure remain to be determined.
The prevailing theory as to CDH development has been that the incomplete closure of the pleuroperitoneal canal was a primarily diaphragmatic defect and that the abdominal viscera herniated to the thorax impeded normal lung development and resulted in the pulmonary hypoplasia and hypertension observed almost universally in association with CDH. This perception was widely confirmed by animal models in which a diaphragmatic defect was surgically produced in fetuses of different species,89–91 or in which the diaphragmatic herniation was mimicked by an inflatable prosthesis placed in the pleural cavity.92 All these models resulted in lung hypoplasia and pulmonary hypertension at birth.
Nonetheless, the foremost notion today is that the primary defect is not in the diaphragm but in the lung buds, and that the diaphragmatic defect would actually be secondary to a primary pulmonary hypoplasia. Such pulmonary hypoplasia, in turn, could be intensified even more by the presence of abdominal viscera herniated into the chest. Starting at the 4th week of gestation, the growth of the pulmonary buds and of the pleural cavities result in a progressive narrowing of the pleuroperitoneal canals and in the formation of the pleuroperitoneal membranes.6,7 The development of the diaphragm, more specifically of the pleuroperitoneal membranes, is intimately related to lung development itself. Researchers that have worked with the experimental model of CDH induced by nitrofen have shown that, in that model, the pulmonary hypoplasia precedes the diaphragmatic defect.82,83,93,94 These findings are in accordance with the fact that nitrofen exposure can lead to pulmonary hypoplasia independently of the existence of CDH.95–97 Iritani has concluded that the lung buds, which are primarily hypoplastic because of exposure to nitrofen, cause hypoplasia of the posthepatic mesenchymal plate, which in turn develops in intimate association with the lung buds.82 The hypoplasia of this mesenchymal plate, which is one of the precursor portions of the primitive diaphragm, would then give rise to the diaphragmatic defect and, consequently, to the CDH.82 Moreover, it is thought that the morphogenesis and differentiation of the pulmonary respiratory epithelium is intimately dependent on the kind of extracellular matrix synthesized by the mesenchyme.83,98,99 Kluth and colleagues have shown that lungs of embryos exposed to nitrofen display an abnormal expression of factors normally found in the extracellular mesenchymal matrix, with a delayed pattern of epithelial differentiation.99 Iritani speculates further that the reason that CDH is more common on the left in humans is that lung bud development tends to be slower on the left than on the right82,100,101 and, also, the fusion of the pleuroperitoneal membranes happens later on the left side than on the right.89 Independent of the possibility of a primary pulmonary hypoplasia in the nitrofen model, Alles and coworkers have shown that there is also cell death in the mesoderm of some cervical somites that are precursors of the diaphragm, suggesting a concomitant primary disorder of diaphragmatic development. Consequently, the mechanism behind the emergence of CDH in this model is still to be clarified.102 Knockout models, such as FOG-2 −/− mice, are further evidence of a primary pulmonary hypoplasia associated with CDH.86
A so-called smooth muscle hypothesis, pointing to abnormalities in airway smooth muscle development as central to the pathogenesis of CD, has been proposed but not yet fully validated,103 and other, less accepted theories for the pathogenesis of CDH exist.73,104–106 Whatever the location and nature of the primary defect may be, the presence of a diaphragmatic defect per se usually leads to herniation of abdominal viscera to the chest, which, in turn, at least contributes to worsen the pulmonary hypoplasia.89–91 The sooner the herniation occurs in gestation and the larger the herniation content, the more intense the pulmonary hypoplasia tends to be.
Pathologic Anatomy
Gross Findings
The diaphragmatic defect, or Bochdalek foramen, is located in the posterolateral aspect of the diaphragm and involves at least the area that would have originated from the pleuroperitoneal membrane and, many times, also the area immediately posterior to it—the lumbocostal triangle (Fig. 33-2). The size of the defect is highly variable, from less than 1 cm in diameter to an almost complete absence of the hemidiaphragm, extending beyond the dome, practically to the midline, preserving merely a small anterolateral band of muscle. There is no fibrosis or any evidence of inflammation at the level of the diaphragmatic opening. The left side is affected in approximately 80% to 90% of the cases, the right side in 10% to 20%, and bilateral cases are rare, occurring in approximately 1% of the patients.17,107

Because of the herniation to the chest, the mediastinum is usually deviated to the contralateral side of the hernia (Fig. 33-3). Both lungs, but particularly the one ipsilateral to the defect, are smaller than normal in both volume and weight (Fig. 33-4).108–110 Pulmonary lobulation is ordinarily normal but may be compromised in a few cases.108,111 On the other hand, the shape of the pulmonary lobes is commonly distorted.108,111 The pulmonary ligament is almost always absent on the side of the hernia.108 The number of airway generations and their dimensions are reduced, especially in the ipsilateral side of the defect.108,111 The pulmonary arteries, as well as the number and dimensions of their branches, are smaller than normal, in proportion to the reduced size of the lungs, and also particularly on the side of the hernia.108
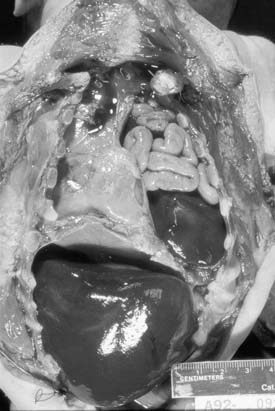
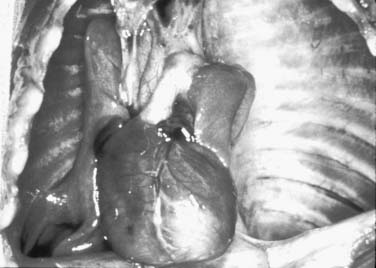
Figure 33–4 Thoracic cavity in the neonate shown on Figure 33-3, after removal of the abdominal organs herniated to the left hemithorax. Note the reduced size of the lungs, especially on the left.
The herniation of abdominal contents to the thorax and the pulmonary hypoplasia may both lead to the appearance of many other abnormalities. Therefore, such abnormalities are not considered other anomalies associated with CDH but actually integral components of the CDH syndrome.112 The most frequent of such abnormalities are persistent ductus arteriosus, persistent foramen ovale, and intestinal malrotation.112 Less frequently, the following abnormalities can also be a direct consequence of CDH: gastric volvulus, abnormally sized chest cavity, accessory spleen or congenital splenic fibrosis in left CDH, abnormal hepatic lobulation or hepatopulmonary fusion in right CDH, and hypoplasia or fibrosis of the lobe of the liver ipsilateral to the hernia.112–114 Also, the volume of the abdominal cavity is frequently reduced.
Microscopy
Except for the defect itself, the diaphragm does not exhibit any other deformity. A local decrease of the density of branches of the phrenic and intercostal nerves has been described by some73 but not yet confirmed by others and is considered of limited value.102
The airway branching order is abnormal, with a reduced number of generations of lower bronchi and bronchioli, often with complete absence of the latter, so that bronchi may end directly in alveoli.108,110,111 Given that normal airway branching is complete by 16 weeks of gestation,100,115 the reduction in airway generations observed both grossly and microscopically is further evidence of the fact that CDH and pulmonary hypoplasia start before this time, more specifically between the 10th and 12th weeks. On the other hand, the development of airway cartilage itself does not seem to be affected, so the proportion of cartilage-bearing airways to the total number of airways is normal.108,111 However, the number of bronchi containing mucous glands is diminished.111
The total alveolar number is reduced, both in absolute terms and in relation to total lung volume.108,110,111 However, the number of alveoli per acinus may be either reduced or normal, suggesting that the reduction in total alveolar number is mostly a consequence of the lower number of terminal bronchioli.108,110 The alveoli are also smaller than normal.108 The fate of type II pneumocytes is not yet completely clear. Many studies uncover evidence that the surfactant system is depressed, but it is not absolutely clear whether this has to do with a reduction in the density of type II pneumocytes.116–122
In parallel to the lower number of airway generations, the absolute number of arterial branches is reduced,108 yet the density of intra-acinar arteries may be normal.110 The arterial diameters are reduced.108,110 There is hypertrophy of the arterial muscle layer at all levels, as well as extension of such muscle layers into more distal branches, which normally would not bear any muscle.108,110,123,124 There seems to be a direct relationship between pulmonary hypoplasia and arterial muscularization, so that the more hypoplastic the lung, the more intense is the abnormally augmented muscularization.123
Pathophysiology
The cardinal aspects of CDH pathophysiology are pulmonary hypertension with persistence of a fetal circulatory pattern, along with a reduction in both pulmonary tidal volume and compliance. The intensity of such manifestations varies a great deal, going from almost nonexistent to incompatible with life, depending mostly on the severity of the anatomic abnormalities of a given patient. According to some, a deficiency of the surfactant system is also part of CDH pathophysiology.116–119 However, this notion has been increasingly challenged by more recent data.122,125,126
In children, total peripheral airway cross-sectional area is proportionally larger than in adults, so the reduction in airway generation present in CDH usually does not lead to significant increases in airway resistance.127 The difficulty in ventilating children with CDH stems mostly from the lower pulmonary compliance and lower tidal volumes. The pressure–volume curves of these hypoplastic lungs are abnormal, so that, at a given pressure, lung volume is lower than normal.128 Microscopic analyses under insufflation show that, although certain airspaces may open at 15 to 20 cm H2O, many are still closed at 30 to 35 cm H2O.129 Thus, higher inspiratory pressures are transferred only to the alveoli that are open, leading to alveolar rupture and a tendency to develop pneumothorax.130 It is not yet clear whether the decreased pulmonary compliance is a result of a quantitative or qualitative (or both) depression of the surfactant system, or is a result a relative increase in the total amount of collagen in the lungs.116–118131 The lower tidal volumes are a direct consequence of the reductions in both lung volume and total alveolar number.108–111 All these ventilatory abnormalities are the main reasons for the tendency of infants with CDH to retain CO2.130
Contrary to what happens with the airways, the peripheral vessels (lower arteries, arterioles, and capillaries) account for most of the pulmonary vascular resistance (PVR). Therefore, as a result of the reduction in the total number of arterial branches and their lower than normal diameters, the total arterial cross-sectional area is diminished and the PVR is usually significantly increased in CDH.132,133 Further contributing factors to the increased PVR are the hypermuscularization of the arteries and amplified arterial reactivity. Because of the latter, certain physiologic stimuli such as alveolar hypoxia, hypoxemia, hypercapnia, acidosis, cyanosis, hypothermia, and any “disturbances,” such as certain inflammatory mediators and simple manipulations of the patient, may trigger intense pulmonary vasoconstriction and marked increase in PVR.134,135 Other than the possibility of a role played by the muscular hypertrophy present in pulmonary arteries and arterioles, the reason these vessels tend to overreact to stimuli is not yet known. Recent data suggest that the pulmonary vasculature’s ability to synthesize nitric oxide is depressed in patients with CDH and thus may be part of the mechanism.136 The possibility of an imbalance involving prostanoids, which are vasoactive agents that include prostaglandins, playing a role has been suggested.133,137–139 A potential role for other endogenous vasoactive agents, such as endothelins, in the increase in PVR has proved debatable.133,140,141
This increase in PVR leads to pulmonary hypertension, which is almost universally observed in neonates with CDH.129,133,142 Pulmonary hypertension leads to a decrease in total blood flow to the lungs, an increase in end-diastolic pressure in the right ventricle, and a tendency for persistence of a fetal circulatory pattern, with right-to-left shunt through the ductus arterious and foramen ovale.129,134 The decrease in total pulmonary blood flow and the right-to-left shunt lead to hypoxemia, hypercapnia, and acidosis, which, in turn, are stimuli to pulmonary vasoconstriction, which worsens the pulmonary hypertension, with consequent intensification of the fetal circulatory pattern and so forth, establishing a vicious circle difficult to break. Not infrequently, patients may be satisfactorily oxygenated and fairly stable, until a random stimulus, or nothing immediately discernible, triggers the vicious circle of fetal circulation. This period of temporary stability that precedes the emergence or worsening of pulmonary hypertension has been coined honeymoon. It was described for the first time by Collins and colleagues in the mid 1970s and remains a frequent observation in neonates with CDH.133,143 The mechanisms responsible for the end of the honeymoon have been elusive until the past few years, when mounting evidence points to it being a cardiac event—more specifically, variable degrees of right heart failure.144 Patients who present soon after birth with persistent hypoxemia, without ever going through a honeymoon period, usually have severe pulmonary hypoplasia and serious abnormalities of the pulmonary vasculature.124
In the fetus, the oxygenated blood that comes from the placenta returns to the right heart through the umbilical vein and crosses either the foramen ovale or the ductus arteriosus toward the aorta, so that only approximately 7% of the cardiac output goes through the lungs.145 Therefore, the hemodynamic disturbances associated with pulmonary hypertension almost never manifest in utero. After birth, however, the hemodynamic status tends to deteriorate, often with overload and potential failure of the right side of the heart. Thus, cardiac failure is typically part of the pathophysiology of CDH,144,146,147 and survival depends, to a great extent, on the ability of the myocardium to withstand the overload imposed by the pulmonary vasculature.129,144,146 The deviation of the mediastinum by the herniated content may lead to a decrease in the venous return to the heart, possibly contributing to further worsening of the patient’s hemodynamic status.
It has been suggested that neonates with CDH may have adrenal insufficiency, with an inadequate response to stress.148 At the same time, there is preliminary experimental evidence pointing to the possibility that lower than normal glucocorticoid levels may contribute to the abnormal lung development and maturation found in patients with CDH.149 The true meaning of these findings in CDH pathophysiology remains to be better defined. Indeed, at least as far as receptors are concerned, hypoplastic lungs of fetuses and newborns with CDH seem to be as responsive to glucocorticoids, thyroid hormone, and retinoic acid (all relevant to normal pulmonary development) as the lungs of normal children.150
Of the different aspects of the pathophysiology of CDH, it is the pulmonary hypertension that is most responsible for mortality in the neonatal period. Because otherwise healthy neonates who undergo total pneumonectomy are often able to maintain good oxygenation and ventilation, without clinically relevant pulmonary hypertension, the lack of pulmonary parenchyma alone cannot explain all the manifestations commonly observed in infants with CDH.151 Rarely in CDH is the bilateral lung impairment intense enough to cause a neonate to have less than half of the normal total alveolar surface area. Consequently, it would appear that, more often than not, the pulmonary vasculature abnormalities are clinically much more relevant than the lack of alveoli (pulmonary hypoplasia).
CLINICAL MANIFESTATIONS
Ninety percent of the patients with CDH are symptomatic within the first 24 hours of life.152 However, this disease may first manifest at any age and, more rarely, go unnoticed until very late in life or even never be diagnosed.153–155
When a child is symptomatic within the first 24 hours of life, the main clinical manifestation is respiratory distress. The earlier the onset of signs and symptoms, the more severe the pulmonary disease. Newborns who are symptomatic in the first 6 hours after birth are considered at high risk and account for 88% of the cases.152 Tachypnea associated with sternal, subcostal, and supraclavicular retraction is common. Cyanosis and pallor are also frequent. Apgar scores tend to be low. If untreated, the dyspnea tends to worsen with time, for three reasons: the progressive distention of the intrathoracic bowel by gas, which is accelerated by aerophagy, common in children in respiratory distress; the gradual increase of the volume herniated to the chest, which is a result of the negative pressure exerted during respiration; and the escalating hypoxemia, hypercapnia, and acidosis, resulting from the vicious circle generated by persistent pulmonary hypertension. The abdomen is often scaphoid because of the migration of abdominal viscera to the chest. However, because of possible bowel distention inside the abdominal cavity, the abdomen may assume a normal appearance with time. The chest may be asymmetrical, larger on the side of the hernia, especially after the bowel fills with gas. The heart sounds are commonly dislocated to the contralateral side of the hernia. Sometimes the same happens with the trachea. In the ipsilateral hemithorax, the respiratory sounds may be diminished or absent altogether, and bowel sounds may be present. There may be hemodynamic instability, with a tendency to arterial hypotension, because of a decreased venous return to the heart resulting from the mediastinal deviation or because of right heart failure due to pulmonary hypertension. Occasionally, mediastinal deviation can also lead to superior vena cava syndrome.156 If untreated, a symptomatic newborn usually expires in a few minutes or hours.
Rarely in the neonatal period, there may be manifestations stemming from perforations or strangulation, or both, of a hollow viscus, gastric, or midgut volvulus, or rupture of a herniated spleen, such as gastrointestinal (GI) obstructions, empyema, hemothorax, fever, arterial hypotension, coagulopathy, anemia, or hypovolemic shock. Anecdotal associations between CDH and septicemia with group B streptococci in premature babies have also been described.157
When CDH first manifests after the neonatal period, partial or complete GI obstructions are more common than respiratory distress, which, if present at all, tends to be mild. Unlike in the neonates, the spectrum of manifestations of late-presentation CDH is quite broad, including, in addition to GI obstruction and respiratory distress, sudden death, growth retardation, perforations or strangulations of intrathoracic hollow viscera (which may lead to sepsis, empyema, pneumothorax, and hemothorax), rupture of a herniated spleen (with hemothorax, anemia, and possibly hypovolemic shock), airway infections or recurrent pneumonias, urinary tract obstruction due to herniation of the ureter, chest pain, abdominal pain, vomiting, diarrhea, anorexia, acute abdomen, intrathoracic appendicitis, and other rare presentations.153,154
In bilateral CDH, both sides may not manifest at the same time, and staggered presentation has been described.158
DIAGNOSIS
The majority of cases are diagnosed before birth, during routine prenatal ultrasound.159 The relative proportion of cases diagnosed in utero is constantly climbing, because of the increasing application of prenatal ultrasound screening and improvements in ultrasound technology and resolution. Fetal ultrasonography should always be performed whenever there is polyhydramnios, as CDH is one of its causes, apparently because of a reduction in the volume of amniotic fluid swallowed by the fetus, probably as a consequence of the GI obstruction caused by the hernia. A few authors also recommend careful ultrasonographic examination whenever an amniocentesis shows abnormally low levels of lecithin and sphingomyelin, because of the possibility of an association between CDH and a deficiency in the surfactant system.118,119 Such an association, however, has been increasingly less accepted.121,122,125,126 CDH can be diagnosed by prenatal ultrasound from the 11th week of gestation until term; previously negative examinations may become positive at any time during the pregnancy.159,160 False-negative and false-positive examinations may occur; fetal ultrasonography is precise in approximately 90% of cases.134,161 Not infrequently, the herniated content identified by prenatal ultrasound moves in and out of the chest, as if the hernia were a dynamic process.162 A case of CDH diagnosed in the second trimester of pregnancy that seemed to have resolved spontaneously during the third trimester, with delivery of a normal infant, has been reported.163 There may be inaccuracies as to the side of the hernia when it is unilateral, and sometimes a bilateral CDH may be diagnosed as unilateral.162 The differential diagnoses of CDH identified by prenatal ultrasound include congenital cystic adenomatoid malformation of the lung (CCAM), diaphragmatic eventration, Morgagni hernia, hiatal hernia, pentalogy of Cantrell, primary diaphragmatic agenesis, pericardial hernia, pulmonary sequestration, lung cysts, diaphragmatic duplication, leiomyosarcoma of the lung, mediastinal teratoma, esophageal atresia with tracheoesophageal fistula, primary pulmonary agenesis, primary pulmonary hypoplasia, and intrathoracic duplications of the GI tract.134,164 Color Doppler, three-dimensional ultrasonography, and magnetic resonance imaging (MRI) may all facilitate the prenatal diagnosis of CDH.165,166 In extremely rare, selected cases, if in doubt, more invasive examinations might be considered, such as amniography, computerized tomography (CT), or ultrasonography with concomitant intrathoracic or intra-abdominal injection of saline as a contrast.162,167 But as MRI becomes more accessible, these more invasive examinations will become of historical interest only. At most referral centers, the prenatal diagnosis of CDH automatically leads to an amniocentesis to determine the fetal karyotype. Should certain chromosomal abnormalities be detected, pregnancy termination may be considered.
After birth, a plain chest radiograph is almost always enough to confirm the diagnosis. The typical image is that of bowel loops seen in the lung field(s), with deviation of the mediastinum to the contralateral side of the hernia, and decrease or absence of gas in the abdomen (Fig. 33-5). When the radiograph is obtained before the GI tract could be filled with gas, or if the intestines are not herniated (which is more common in right hernias), there may be confusion in the diagnosis. The introduction of a radiopaque gastric tube often helps, in case the stomach is herniated (see Fig. 33-5). Should any uncertainty persist, which is highly uncommon, the diagnosis can be confirmed through a radiograph performed after infusion of contrast through the gastric tube. Even less frequently, an ultrasound may be of help. More rarely, a CT, MRI, or contrast enema may have a role.
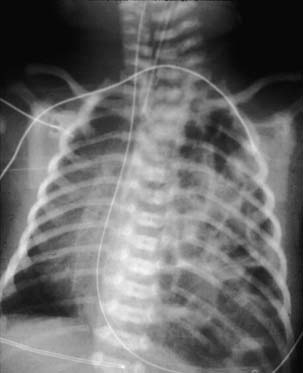
For late-presentation CDH, the diagnosis is usually made through a simple chest radiograph as well. Also in these cases, a gastric tube may be of help during the interpretation of the radiograph. Many times, there is previous history of a normal chest radiograph.168 The possibility of late-presentation CDH should always be considered, to avoid delay or confusion with either pneumonia, pneumatoceles, CCAM, pneumothorax, pleural collections, diaphragmatic eventration, lung cysts, lung nodules, or pulmonary sequestrations. Because late-presentation CDH is relatively rare, the need for other imaging in addition to the chest radiograph is somewhat more common. Such imaging may include an upper GI series (often with the patient in Trendelenburg position), ultrasound, CT, MRI, fluoroscopy, and, more rarely, a contrast enema. CDH may be diagnosed as an incidental imaging finding in an asymptomatic patient.169
ASSOCIATED ANOMALIES
Children bearing any major congenital anomaly are known to carry a much higher risk of having another anomaly than the general population, and neonates with CDH are no exception. The incidence of other anomalies associated with CDH in the literature varies from “rare” to as much as 60%.54,56,112,170,171 Explanations for this disparity include other anomalies directly linked to CDH and considered integral components of the so-called CDH syndrome, as mentioned earlier; stillbirths or patients who die before reaching a referral center; and variabilities in diagnostic routines, autopsy rates, patient populations in terms of the proportion of high-risk neonates included in the analysis, and regional prevalences of certain congenital anomalies. Studies have identified many different associated anomalies, in all body systems, usually with a predominance of cardiac malformations.
In a detailed review of 166 high-risk neonates (i.e., symptomatic within the first 6 hours of life), we noticed that approximately 40% of the children had one or more congenital anomalies associated with CDH.112 This index was obtained even after exclusion of all the other anomalies that are part of the CDH syndrome. Cardiac anomalies were by far the most common, found in 63% of the patients who had an associated anomaly, followed by anomalies in the genitourinary tract (23%), GI tract (17%), central nervous system (CNS) (14%), muscles and skeleton (10%), chromosomes (10%), lungs (5%), and others (5%); in many children, there were more than one associated anomaly (Table 33-2).112 Our results were comparable to those from another large series.54,171
Table 33–2 Associated Anomalies Indentified in 166 High-Risk Patients with Congenital Diaphragmatic Hernia∗
| Anomaly | Patients (N) |
|---|---|
| Cardiac | |
| Heart hypoplasia | 13 |
| Atrial septal defect | 10 |
| Ventricular septal defect | 9 |
| Hypoplasia of aortic isthmus | 3 |
| Aortic coarctation | 3 |
| Persistent left superior vena cava | 3 |
| Ebstein’s anomaly | 2 |
| Parachute mitral valve | 2 |
| “Abnormal” mitral valve | 1 |
| “Abnormal” tricuspid valve | 1 |
| Absent left pericardium | 1 |
| Absent right pulmonary artery | 1 |
| Bicommissural aortic valve | 1 |
| Bifid apex of the heart | 1 |
| Common atrioventricular canal | 1 |
| Cor triatriatum | 1 |
| Double outlet of right ventricle | 1 |
| Double coronary ostia | 1 |
| Scimitar syndrome | 1 |
| Single coronary artery | 1 |
| Genitourinary | |
| Undescended testes | 6 |
| Bicornuate uterus | 2 |
| Hydronephrosis | 2 |
| Horseshoe kidneys | 1 |
| Hypospadias | 1 |
| Renal dysplasia | 1 |
| Single kidney | 1 |
| Ureteropelvic junction obstruction | 1 |
| Vaginal or uterine atresia | 1 |
| Central Nervous System | |
| Hydrocephalus | 4 |
| Rachischisis | 2 |
| Circle of Willis anomaly | 1 |
| Microcephaly | 1 |
| Myelomeningocele | 1 |
| Open spine | 1 |
| Vascular malformation of cord | 1 |
| Gastrointestinal | |
| Meckel’s diverticulum | 6 |
| Absent gallbladder | 1 |
| Absent vermiform appendix | 1 |
| Accessory pancreas | 1 |
| Annular pancreas | 1 |
| Duodenal atresia | 1 |
| Ectopic liver | 1 |
| Ectopic pancreas | 1 |
| Esophageal atresia with transesophageal fistula | 1 |
| Imperforate anus | 1 |
| Neuroenteric cyst | 1 |
| Phrygian cap deformity of gallbladder | 1 |
| Musculoskeletal | |
| Hemivertebrae | 2 |
| Absent rib | 1 |
| “Abnormal” rib | 1 |
| Accessory rib | 1 |
| Hip dislocation | 1 |
| “Limb dystrophy” | 1 |
| Polydactyly | 1 |
| Sacral dysgenesis | 1 |
| Scoliosis | 1 |
| Chromosomal | |
| Trisomy 18 | 2 |
| “Abnormal” chromosome 14 centromere | 1 |
| Balanced 12/15 translocation | 1 |
| Chromosome 7Q deletion | 1 |
| Chromosome 12P | 1 |
| Mosaic trisomy | 1 |
| Tetraploidy 21 | 1 |
| Trisomy 13 | 1 |
| Pulmonary | |
| Pulmonary sequestration | 2 |
| Pulmonary lymphangiectasia | 1 |
| Trifurcated trachea | 1 |
| Other | |
| Inguinal hernia | 2 |
| Omphalocele | 2 |
| Cleft lip or palate | 1 |
| Conotruncal facies | 1 |
| Torticollis | 1 |
∗ Excluding pulmonary hypoplasia, persistent ductus arteriosus, persistent foramen ovale, intestinal malrotation, gastric volvulus, size abnormalities of the chest wall, accessory spleen or congenital splenic fibrosis in left congenital diaphragmatic hernia (CDH), abnormalities of liver lobulation in right CDH, and hypoplasia or fibrosis of the liver lobe ipsilateral to the hernia.
Reproduced with permission from Fauza DO, Wilson JM. J Pediatr Surg 1994;29:1113-7.
The high proportion of cardiac anomalies associated with CDH in many series deserves special attention. In our review, for example, they were more frequent than all other anomalies put together (see Table 33-2).112 Among the cardiac anomalies, heart hypoplasia was the most common.112 This finding is in accordance with many observations pointing to the fact that the cardiac hypoplasia associated with CDH, especially that of the cardiac chambers ipsilateral to the hernia, occurs, like the pulmonary hypoplasia itself, at least in part because of compression of the heart by the herniated content and, perhaps, should also be considered part of the CDH syndrome, as it probably plays a role in the cardiac failure often manifested clinically.112,172–174 The presence of an associated cardiac anomaly may lead to significant further reduction of the postductal PO2 when compared with isolated CDH.112 In fact, in neonates with an excessively low postductal PO2, a potential associated cardiac anomaly should be searched for very carefully.
The side of the hernia has recently been linked to differences in the frequency and pattern of associated malformations.175 This interesting first observation needs further validation. Patients with late-presentation CDH also seem to have a higher prevalence of other anomalies than the general population. However, the related series published thus far are too small for definitive conclusions. For example, in a review of 26 patients, spanning 20 years, Berman and coworkers found one or more associated anomalies in 31% of them.176
PROGNOSTIC FACTORS
Certain clinical variables are known to be associated with different survival rates. Ever since the study by Young, in 1969, there is a well-established inverse relationship between age at the beginning of symptoms and mortality rates.55,177 Neonates symptomatic within the first 6 hours of life are considered high risk, as they have the lowest survival rates.55 There is no difference in mortality between the sexes in isolated CDH, but higher mortality rates have been reported in females than in males, when CDH is associated with other anomalies.54 Some studies have shown that the lower the gestational age at birth, or the lower the birth weight, the lower is the survival rate.55,178 Lower Apgar scores, particularly in the 5th minute, are also linked to higher mortality.55 Contrary to what a few studies have suggested, the side of the hernia has not had any prognostic impact in the larger series.55 At the same time, the size of the defect is well established as a determining factor in survival.24,55 It is not yet known whether the presence of a hernia sac is of any prognostic significance. The variable with the highest impact on CDH survival rates is the presence of other associated congenital anomalies. The prognosis for neonates with associated anomalies, especially cardiac, is worse than that of infants with isolated CDH.55,112,179,180
Since the beginning of the 1970s, countless studies have tried to correlate mortality with blood gas values or ventilation parameters, either independently or mutually integrated in often intricate equations. Examples include pH values; “best” PCO2 or “best” PO2, either postductal or preductal; alveolar–arterial O2 gradient; mean airway pressure; respiratory rate; pulmonary compliance; dead space; and tidal volume.181–186 In the past, perhaps the most popular of these markers was the best postductal PO2 obtained during maximal mechanical ventilation, either prior to surgical repair of the hernia or prior to placing the patient on ECMO. Children with values higher than 100 mm Hg were labeled “responsive” and had a better prognosis, and vice versa.184,185 Other parameters also used until recently were the so-called Bohn criteria, which related PCO2 with the ventilation index (VI = respiratory rate × mean airway pressure); for example, a PCO2 greater than 40 mm Hg with a VI of 1000 or greater would suggest high mortality.182,184,185 Some authors have proposed that preductal blood gases are more predictive of the degree of pulmonary hypoplasia than postductal blood gases, as the latter are more influenced by the intensity of pulmonary hypertension and right-to-left shunt; a preductal PO2 of less than 100 mm Hg and a preductal PCO2 of greater than 60 mm Hg would be related to very high mortality.183,187 As a result of the now universal acceptance of the principle of gentle ventilation, permissive hypercapnia, and minimization of iatrogenic injury related to mechanical ventilation, even preductal blood gases have had increasingly limited prognostic value, and at the same time, postductal gases such as best PO2 during maximal ventilation have been practically abandoned.
Imaging criteria as severity predictors have also been extensively studied. One example is the value of prenatal ultrasonography. Until the early 1990s, a positive prenatal ultrasound was considered a marker of bad prognosis, particularly if the hernia was diagnosed before 25 weeks of gestation.188,189 Lately, probably as a result of the widespread use of fetal ultrasound during routine prenatal care, the improvements in ultrasound technology, and the novel therapeutic strategies for CDH, it is quite clear that the prenatal diagnosis of an isolated CDH is, by itself, of no prognostic value, regardless of the gestational age at which it is made.159,179 In the event that other associated anomalies, especially cardiac, are diagnosed prenatally in addition to the diaphragmatic hernia, the prognosis is still comparable to that of a diagnosis made after birth.112 Several specific findings on fetal ultrasound have been proposed as bad prognostic markers, such as polyhydramnios, herniation of the liver or the stomach into the chest, underdevelopment of the left side of the heart, disproportionate cardiac ventricles, high ratio of the herniated area to the cardiac area, low ratio of the lung area to the total thoracic area, depression or absence of fetal breathing movements, severe mediastinal deviation, reduction of liquid flow through the nose and oropharynx during fetal breathing movements, and disturbances of blood flow modulation through the ductus arteriosus.160,188–191 The merit of all these markers, however, is highly controversial and of very limited acceptance. Somewhat recently, the value of liver herniation, as well as of the so-called lung-to-head ratio (LHR), which measures the relative proportion between the areas of the lung and the head at predetermined locations, has been emphasized by a few groups.192,193 For example, fetuses with liver in the chest and an LHR of less than 1 would have a particularly bad prognosis. Although debate continues over the value of the presence of the liver in the chest, the same cannot be said of the LHR, which has been shown to be significantly inconsistent, especially across different institutions.152,194 Three-dimensional measurements of fetal lung volume via MRI are being increasingly refined and adopted as a more reliable imaging-based prognostic marker, including at our institution.165,195
Similarly, some studies suggest that certain postnatal imaging findings, either independently or in combination, can also be linked to poor outcome. On plain chest radiograph, examples of such findings include presence of the stomach in the chest, ipsilateral or contralateral pneumothorax, presence of interstitial emphysema, and a low ratio of aerated ipsilateral lung area over that of the contralateral lung.196–198 On echocardiogram, examples are decreased left ventricular mass, disproportionate dimensions between both pulmonary arteries, and a disproportionately large pulmonary artery trunk in relation to the aorta.199,200 Finally, on pulmonary arteriogram, examples are reduced dimensions of both pulmonary arteries, reduced size of the ipsilateral lung, and severe peripheral compromise.184 Yet, as with prenatal imaging, the predictive value of these proposed postnatal findings is questionable, to say the least, and very few institutions adopt any of them at this time.
Only the following variables have been clearly validated by well-controlled, extensive multicenter data as being of predictive value in CDH: age at the onset of symptoms, birth weight, 5-minute Apgar score, defect size, and the presence of an associated cardiac anomaly.24,55 The meaning of all other suggested prognostic markers is debatable and of limited acceptance.201 The impact of various prognostic markers seems to differ depending on the side of the hernia.202 One of the most common shortcomings of the studies involving prognostic markers is that most of them are reviews from a single institution. Hence, the data are unavoidably linked to the unique patient population and peculiar therapeutic strategies of each service, which are aspects known to still be highly variable from one center to another in CDH. On the other hand, even if well-controlled multicenter trials were performed, the fact that the treatment of CDH is constantly evolving could lessen the significance of their results. The introduction of ECMO and of the principle of gentle ventilation are clear examples of the vulnerability of data of this kind, as these therapies rendered the conclusions obtained prior to their availability nearly useless, even in a given institution.
TREATMENT
Except for the rare cases of strangulation of the herniated content, CDH is not a surgical emergency. Rather, CDH is a physiologic emergency. In fact, not infrequently, mechanical aspects of respiration tend to deteriorate after the repair of the hernia.203,204 Indeed, not only is emergency surgery unnecessary and often deleterious, but also a period of preoperative stabilization is known to improve outcome.22,205 The child should not go to the operating room while unstable. The time needed for stabilization may vary from less than 12 hours to several days. A few authors even suggest that waiting until well beyond stabilization has been reached is beneficial.22 In certain premature neonates at higher surgical risk, one may wait weeks, or even more than a month, before proceeding to the repair.152
The therapeutic strategy in children with CDH and other associated anomalies must be individualized for the patient. Usually, the hernia is repaired before the other anomalies. However, when CDH is associated with cardiac anomalies, such strategy may lead to unacceptably high mortality.112,152 Unless the cardiac defect is very mild, the current tendency is to repair the heart before the diaphragm, or, occasionally, both during the same intervention. However, guidelines for the different scenarios are still being defined.
One of the benefits of the prenatal diagnosis of CDH is that delivery can be planned at a tertiary referral center. The initial results of postnatal resuscitation are known to be maximized when the presence of CDH is detected before birth.159 At our institution and others, high-risk outborn infants have a significantly lower survival rate than inborn neonates. Unless there is any obstetrical contraindication, vaginal delivery is preferred over cesarean section.
Preoperative Care
The neonate should be intubated in the delivery room. Ventilation by mask before intubation should be avoided because of the risk for distention of hollow viscera in the chest. A gastric tube should be introduced and kept under mild continuous suction, to minimize such distention and to drain air that may have been swallowed. Central venous access is established, usually through the umbilical vein (occasionally, however, particularly in patients with right-sided CDH, there may be significant distortions of the liver anatomy because of the herniation, which may impede the use of this vein). One of the umbilical arteries is catheterized for blood pressure and postductal blood gas monitoring. Preductal blood gases are obtained through access to the right radial artery, or to one of the superficial temporal arteries. Transcutaneous pulse oximetry monitors and, if available, transcutaneous PO2 and PCO2 monitors are placed both on preductal and postductal territories. Monitors for body temperature and respiratory rate should also be positioned. At least in the high-risk cases, a Foley catheter is introduced. The volumes of intravenous infusions should be carefully controlled to minimize the chances of a pulmonary edema developing. Prophylactic antibiotics covering both gram-positive and gram-negative bacteria are commonly administered. Whenever possible, inotropic agents should be avoided, as these drugs usually increase not only cardiac output but also peripheral vascular resistance, both of which may, together with the ever-present pulmonary hypertension, lead to excessive cardiac overload not always tolerated, especially when there is some degree of cardiac hypoplasia.112,173 When the use of these drugs is inescapable, many prefer dobutamine, amrinone, or epinephrine to dopamine or norepinephrine, because the former drugs may produce pulmonary vasodilation at low dosages.134 One should limit any manipulation or interaction with the infant to a minimum because of the great volatility of the pulmonary vasculature.
Every neonate with CDH should undergo an echocardiogram with Doppler, given the relatively common occurrence and prognostic impact of both right heart failure and associated cardiac anomalies. Should any functional or structural cardiac disease be identified, its treatment must be carefully coordinated with that of the hernia. Not infrequently, a pediatric cardiologist is a member of the multidisciplinary team caring for these patients. Certain variables, such as prenatal diagnosis prior to the 25th week of gestation, low Apgar scores, and “excessively low” postductal PO2, have been shown to be risk factors for the presence of other associated anomalies.112 Should these variables be present, further examinations such as genitourinary tract and head ultrasounds, as well as a karyotype (if this was not already done during pregnancy) should be strongly considered, in addition to the echocardiogram.112
Until the early 1990s, neonates were typically sedated, paralyzed, and hyperventilated, and they also received systemic alkalinization with sodium bicarbonate or tromethamine to minimize pulmonary hypertension. Ventilation parameters used to be controlled by postductal blood gases. This strategy, which unfortunately is still practiced by many centers today, is clearly associated with unacceptably high risks for iatrogenic lung injury. Pulmonary hypoplasia and, in particular, pulmonary hypertension are both expected to lead to low PO2 and high PCO2, especially in postductal gases. Attempts to normalize postductal blood gas values often lead to marked increases in ventilator parameters—namely, respiratory rate (RR), inspired O2 fraction (FiO2), peak inspiratory pressure (PIP), positive end-expiratory pressure (PEEP), and mean airway pressure (MAP), which, in turn, commonly lead to hyperdistention of the lungs and severe barotrauma, not to mention the toxicity of high FiO2 levels. A survey involving clinical and autopsy data from 68 children with CDH treated in this fashion showed a tremendously high frequency and severity of iatrogenic insult to the pulmonary parenchyma.174 In that study, 91% of the patients had evidence of diffuse alveolar damage with development of hyaline membrane, more obvious in the ipsilateral lung. Moreover, 65% of the children developed pneumothorax, 51% had pulmonary hemorrhage, and 6% already had variable degrees of interstitial fibrosis.174 Other studies also showed alveolar ruptures, damage to the alveolar basal membrane, alveolar hemorrhage, and edema, all of which contributed to atelectasis, a decline in lung compliance, and even further deterioration of gas exchange.16,122 In addition to barotrauma, to which CDH neonates are particularly vulnerable,129 pulmonary hyperdistention leads to even further increases of the already elevated pulmonary vascular resistance, thus worsening the effects associated with pulmonary hypertension.206,207 Systemic alkalinization with sodium bicarbonate may lead to increases in PCO2 and to both volume and sodium overloads. Tromethamine may be useful at times in the short term, but relatively large volumes of this drug are usually necessary, which tends to result in both generalized and pulmonary edema.
It is very clear that a completely different strategy, first proposed by Wung and colleagues, should be offered to high-risk neonates with CDH.21,22 It is based on the following guidelines: minimal sedation; no muscle paralysis; respiration merely assisted by the mechanical ventilator, if possible through pressure support under flow synchronization, or, if this is not available, then through simple or synchronized intermittent mandatory volume; permissive hypercapnia with no hyperventilation; and no systemic alkalinization. Patient monitoring is mostly through preductal gases. Ventilator parameters are left at the minimum necessary to maintain a preductal arterial O2 saturation (SaO2) of 90% or greater, whatever the postductal values may be, with tolerance to high PCO2 levels. In general, the ventilator parameters are left at a base RR of 40 breaths or fewer per minute, and the PIP and PEEP no higher than 30 and 5 cm H2O, respectively, and the FiO2 should be the lowest possible to prevent severe preductal hypoxemia. That is to say, unless there is metabolic acidosis, suggesting excessively low O2 delivery, one should tolerate low postductal PO2 and SaO2, as long as there is an adequate amount of oxygen in the preductal blood that is going to the brain and to the heart. As long as the pH is at “acceptable” levels, one should tolerate hypercapnia. The main goal of this therapeutic strategy is the prevention of barotrauma, probably the most common cause of death whenever the old, conventional hyperventilation strategy is employed.16,22,187 Despite the known benefits of sedation in patients with CDH, which raises the excitability threshold of the pulmonary vasculature,208 in this “new” strategy, one must use sedation with great caution, so that a child can effectively activate the mechanical ventilator, preferably in pressure support under flow synchronization. A child who is able to control both the RR and the airflow from the respirator can contribute more to the minute volume without compromising the functional residual capacity. This therapeutic strategy also avoids the side effects of systemic alkalinization.
Pulmonary Vasodilators
Regardless of the ventilation strategy, many vasodilators administered systemically have been used in an attempt to control the pulmonary hypertension. Examples of these agents are tolazoline, nitroglycerin, nitroprusside, acetylcholine, prostaglandin E1, prostaglandin D2, prostacyclin, isoprenaline, and nifedipine. Despite the theoretical appeal of these drugs, they are not selective enough to the pulmonary vasculature and usually lead to a drop in both total peripheral vascular resistance and systemic arterial pressure. Therefore, their effects on the pressure gradient through the ductus arteriosus is either minimal or nonexistent, so that the tendency to right-to-left shunt is unchanged. Furthermore, they may lead to vasodilation of poorly ventilated areas of the lungs, which may even worsen the intrapulmonary right-to-left shunt. The drop in systemic arterial pressure, on the other hand, may lead to the administration of volume or inotropic agents (or both), both of which should be avoided. Bos and coworkers reported a reduction of both the alveolar–arterial O2 gradient and the oxygenation index (OI = MAP × FiO2/PO2) after administration of prostacyclin in high-risk CDH patients, but this had no impact on survival.209 The use of prostacyclin is also associated with an increase in bleeding time, which is clearly undesirable in surgical patients.209,210 Inhalational prostacyclin has been investigated experimentally but has found no clinical applicability yet.211,212 The response to prostaglandin D2 is variable, and systemic hypotension is a common side effect.133,209,212 The most popular of these vasodilators was, perhaps, tolazoline, an α-adrenergic receptor blocking agent with a mild inotropic and chronotropic effect on the myocardium. This drug also lowers the levels of thromboxane B2, which is possibly a mediator of pulmonary hypertension in CDH and which also has histamine-like effects.134,213 Children who respond to tolazoline usually do so within 4 hours after an initial bolus of 1 to 2 mg/kg, which may be noticed by an increase in the postductal PO2. This bolus is commonly followed by a continuous infusion of 1 mg/kg/hr. The infusion of vasodilators directly into the pulmonary artery has been shown not to have any advantage over their systemic or peripheral administration.214 Tolazoline side effects may be severe and include systemic arterial hypotension, upper GI hemorrhage, thrombocytopenia, hyponatremia, and skin rubor.134,213 If there is upper GI hemorrhage, one should give priority to antacids and gastric lavage, as tolazoline may inhibit the effects of cimetidine.215 Tolazoline, as well as these other vasodilators, may occasionally contribute to the stabilization of the patient, sometimes lengthening the honeymoon period. However, more often than not, this is not the case and, even when there is some improvement in oxygenation, no beneficial impact on survival has been demonstrated.22,205,209 Indeed, most referral centers no longer use systemically administered pulmonary vasodilators.
More recently, sildenafil has been proposed as beneficial in the management of pulmonary hypertension associated with CDH.216–218 Although the initial reports are arguably encouraging, its therapeutic value remains to be defined.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


