Acyanotic
• Left-to-right shunts
Atrial septal defects
Ventricular septal defects
Atrioventricular septal defects
Aortopulmonary window
• Left-sided obstructive lesions
Aortic coarctation
Congenital aortic stenosis
Interrupted aortic arch
Cyanotic
• Right-to-left shunts
Tetralogy of Fallot
Pulmonary stenosis
Pulmonary atresia
– With intact ventricular septum
– With ventricular septal defect
Tricuspid atresia
Ebstein’s anomaly
• Complex mixing defects
Transposition of the great vessels
Total anomalous pulmonary venous connection
Truncus arteriosus
Hypoplastic left heart syndrome
In the relative short history of surgical treatment for congenital heart defects, there has not been a more important technological breakthrough than the heart-lung bypass machine. Understanding of the various methods of its use is instrumental in understanding the logistics of surgical treatment of congenital heart disease.
10.2 Cardiopulmonary Bypass
Cardiac function is critical to the maintenance of ongoing hemostasis for any organism. Often, repair of congenital heart lesions requires interruption of this function for brief periods of time. The cardiopulmonary bypass circuit provides systemic support during these periods of cardiac arrest. A standard approach in cardiopulmonary bypass is draining blood from the right atrium and delivering it to the aorta past the aortic valve, with blood being oxygenated within the bypass machine. Blood not actively captured in the right atrium is sent through the lungs and out of the left ventricle when the heart is still beating. By directing the venous drains into the superior and inferior cavae and then snaring down on the cannulas, the right atrium can be safely entered as long as there is no communication with the left side of the heart (atrial septal defect, ventricular septal defect, etc.). Some lesions on the right side of the heart can therefore be fixed while the heart is still beating.
Alternatively, lesions that communicate with or are within the left side of the heart need an alternative strategy to prevent air being passed into the cranial vessels. To isolate the left heart, a clamp is placed between the aortic valve and the outflow of the cardiopulmonary bypass cannula. The heart is placed in a metabolically protected state by both physically cooling the heart and chemically inactivating it with a solution of cardioplegia. The length of time that the clamp can stay on depends on the solution used and the baseline function of the heart.
A third method in operating on the heart is to metabolically inactivate the entire body through cooling, therefore not needing any cardiopulmonary function for a short period of time. Cardioplegic arrest is used in procedures where maintaining the outflow of the bypass machine is not possible. In this method, the bypass machine is used to cool the entire body to a level where the period of decreased flow will not lead to permanent damage. The outflow of the bypass machine is turned off once the body has been sufficiently cooled and work is done expeditiously. Through these various bypass methods, the many varied congenital heart defects can be surgically treated. The remaining portions of the chapter will describe treatment methods used for specific lesions. For additional information, see Chap. 33.
10.3 Systemic Venous Anomalies
Persistence of the left-sided superior vena cava (LSVC) in isolation can be a variant of normal, when it drains to the coronary sinus, which is usually of no physiologic consequence. When an LSVC drains directly to the left atrium, or to an unroofed coronary sinus, systemic venous blood mixes with the pulmonary venous return, causing systemic desaturation. A persistent LSVC may herald additional anatomical issues or be part of a complex of lesions seen in heterotaxy syndromes. When an LSVC is paired with congenital heart lesions, this may alter the repair and must be identified and taken into consideration in preoperative planning [3].
The term interrupted inferior vena cava (IVC) describes the absence of the intrahepatic IVC, and the IVC blood is diverted to the superior vena cava (SVC) via the azygos vein. This finding is of physiologic significance in patients who need to undergo single ventricle palliative procedures in which the SVC blood is diverted to the pulmonary arteries (cavo-pulmonary or Glenn anastomosis), followed years later by diversion of the IVC blood to the pulmonary arteries (Fontan procedure). In this situation, the large majority of the systemic venous blood will be channeled to the pulmonary arteries with the Glenn anastomosis, which is not well tolerated at a young age, and different strategies must be employed. In the setting of an interrupted IVC, a full evaluation for other indicators of a heterotaxy syndrome must be performed [3].
10.4 Atrial Septal Defects
Fetal circulation requires the presence of a patent foramen ovale (PFO), or communication between the right and left atria, for the more highly saturated umbilical venous blood to bypass the lungs and be directed to the brain. After birth, pulmonary blood flow increases, closing the flap of the foramen ovale in approximately 75 % of all individuals, with the remainder maintaining at least probe patency of this communication [4]. This small communication is most commonly of no hemodynamic significance in early life.
Secundum-type atrial septal defects (ASDs) are due to deficiencies in the septum primum portion of the atrial septum, which allows a portion of the pulmonary venous return to cross the atrial septum into the right atrium, then into right ventricle, and finally recirculate to the lungs. When large enough, this leads to dilation of the right atrium and ventricle, and over time, this overcirculation of the lungs can lead to permanent damage of the pulmonary arterial vasculature and/or lung disease [5]. Primum type ASDs will be discussed in the atrioventricular septal defect section. Sinus venosus-type ASDs include defects in the septum separating the cavae from the pulmonary venous return. This allows for drainage of the associated pulmonary venous return to the right atrium, with the same hemodynamic consequences as a secundum-type ASD. This will be further discussed in the section on pulmonary venous anomalies.
Coronary sinus ASDs present variably, including as a partial or completely unroofed coronary sinus, i.e., allowing desaturated blood to drain to the left atrium and left atrial blood to cross the os of the coronary sinus into the right atrium. This can associated with a persistent LSVC (effectively draining directly to the left atrium), with additional atrial septal communications, or with atrioventricular valve abnormalities [6]. The physiologic consequences of this communication and shunting are similar to a secundum-type ASD (Fig. 10.1).
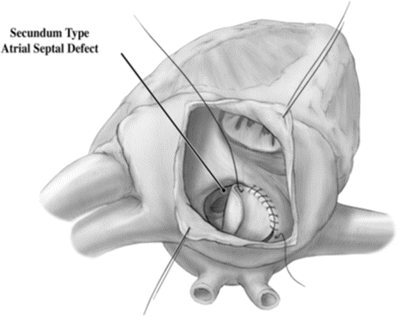
Fig. 10.1
Secundum-type atrial septal defect seen via an incision in the right atrial appendage
The correction of an ASD involves closing off the communication between the right and left atria. Correction is usually performed as a young child prior to long-term sequelae of a left-to-right shunt development. Surgical correction requires an arrested heart with a bicaval cannulation. As there are many types of ASDs, there are different surgical corrections. The most standard defect, the secundum defect, can be closed by simply closing the defect with suture or, if larger, using a patch material to close the defect. Traditionally the patient’s own pericardial tissue has been used as patch material, although pericardium from animals or completely artificial materials such as GORE-TEX can be used. Closure of sinus venosus generally requires more extensive redirection of blood flow to allow proper drainage of the pulmonary veins to the left atrium. In these cases a patch must baffle the blood flow of the pulmonary veins to the left atrium. Pulmonary venous connection with the SVC requires disconnecting the portion of SVC communicating with the veins and using the right atrial appendage as a graft to the remaining SVC in a so-called Warden procedure.
10.5 Anomalies of the Tricuspid Valve
Tricuspid atresia refers to a complete lack of communication between the right atrium and right ventricle and can present in multiple forms. The inlet portion of the right ventricle is underdeveloped, with the remainder of the ventricle dependent on the presence and size of a ventricular septal defect and the ventriculo-arterial connection. Table 10.2 describes the various subtypes of tricuspid atresia, organized by relationship of the great vessels, with type I referring to normally related great vessels, type II describing d-transposed great vessels, and type III as l-transposed great vessels; subtypes describe the amount of pulmonary stenosis (or atresia) [7].
Table 10.2
Tricuspid atresia
Normally related great vessels • Type I (a) pulmonary atresia • Type I (b) pulmonary hypoplasia, small VSD • Type I (c) no pulmonary hypoplasia, large VSD | |
D-transposed great vessels • Type II (a) pulmonary atresia • Type II (b) pulmonary or subpulmonary stenosis • Type II (c) large pulmonary artery | |
L-transposed great vessels • Type III (a) pulmonary or subpulmonary stenosis • Type III (b) subaortic stenosis |
Ebstein’s anomaly is a spectrum of anomalies of both the tricuspid valve and right ventricle, with displacement of the tricuspid valve annulus toward the apex of the heart. The tricuspid valve is adherent to the right ventricle, prohibiting complete coaptation or closure of the tricuspid valve leaflets. This leads to regurgitation of the tricuspid valve and resultant dilation of the right atrium and ventricle. An ASD, pulmonary stenosis or atresia, and/or Wolf-Parkinson-White syndrome may often be associated with this anomaly [8]. In 1988, Carpentier and colleagues attempted to classify Ebstein’s anomalies based on the relative volumes of the right ventricle. More specifically, these authors proposed four subtypes: (1) type A, the volume of the right ventricle is adequate; (2) type B, there is a large atrialized component of the right ventricle, but the anterior leaflet moves freely; (3) type C, the anterior leaflet is severely restrictive in its movement and may cause significant obstruction of the right ventricular outflow tract; and (4) type D in which there is almost complete atrialization of the ventricle, with the exception of a small infundibular component.
Surgical repair of an Ebstein’s anomaly is dependent on the degree and timing of presentation of tricuspid insufficiency. Neonates with severe tricuspid insufficiencies that inhibit forward flow through the right heart will need early interventions due to severe cyanosis; this approach typically leads neonates to a Fontan circulation. The first stage is performed as a neonate with an arrested heart. In addition to the creation of an aortopulmonary shunt, the tricuspid valve is patched closed, and the atrial septum is resected.
In patients that have less severe tricuspid regurgitation, repair is delayed until symptoms develop. Surgical goals are to form a well-functioning tricuspid valve and close any atrial septal communication. Many techniques have been developed, most of which involve plicating and/or resecting the atrialized portion of the right ventricle.
10.6 Ventricular Septal Defects
Ventricular septal defects (VSDs) are channels that permit interventricular shunting and can be found in isolation or in association with other congenital malformations. These channels may be described by their locations within the interventricular septum that is deficient, or by their anatomical nature [9], and also assigned a size relative to other anatomical structures of the heart (most often the aortic valve). The physiologic sequelae of VSDs depend on the size and location of the defects, with unrestricted VSDs leading to elevations of the right ventricular and pulmonary arterial pressures. Further, over time, the pulmonary vasculature develops muscular thickening which, if left unchecked, can lead to irreversibly high pulmonary vasculature resistance, or Eisenmenger’s syndrome.
Redundant nomenclature exists for VSDs in the literature (Table 10.3). The intraventricular septum can be divided into four quadrants as seen in Fig. 10.2 [10] utilizing the septomarginal trabecularis as a landmark. For each region of the IVS, there are unique concerns and probabilities of spontaneous closure of a defect. Defects posterior to the posterior arm of the septomarginal trabecularis, inferior to the atrioventricular valve, are termed inlet or atrioventricular canal-type VSDs and are further discussed in the atrioventricular septal defect section. Defects of the trabecular or muscular septum are the most likely to close spontaneously and can be subcategorized into anterior, posterior, mid-ventricular, or apical [10]. Those defects within or in close proximity to the membranous septum are termed perimembranous, paramembranous, or conoventricular VSDs. Defects in the outlet regions of the right ventricle (superior to the anterior portion of the septomarginal trabecularis) carry the names subarterial, supracristal, conal, or infundibular. These defects can lead to prolapse of the aortic valve and thus aortic regurgitation [11]. When closing these defects surgically or with a device, one must remain cognizant of the conductive tissues that pass in close proximity to many of these defects.
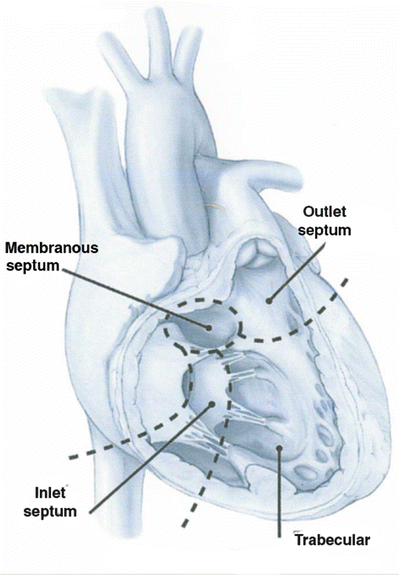
Table 10.3
Classification of ventricular septal defects
Type 1 | Subarterial Supracristal Conal Infundibular |
Type 2 | Perimembranous Paramembranous Conoventricular |
Type 3 | Inlet Atrioventricular canal type |
Type 4 | Muscular • Anterior • Midventricular • Posterior • Apical |
Fig. 10.2
Ventricular septum visualized through the right ventricular free wall. Note the three regions, including the inlet region supporting the tricuspid valve, the trabeculated muscular septum, and the outlet septum forming the pulmonary annulus
The correction of VSDs involves closing off the communications between the right and left ventricles. Corrections are usually performed in infants, as heart failure symptoms can develop that then, in turn, limit growth.
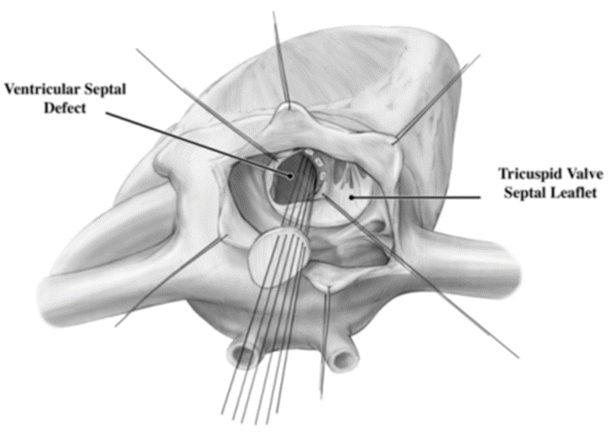
Surgical correction of VSDs typically requires an arrested heart with a bicaval cannulation. A correction almost always requires a patch, as the stresses on the repair site are much higher than that on an ASD (Fig. 10.3). The patch often will abut the conduction system which, if damaged, may lead to the lifelong need for an implanted pacemaker, i.e., secondary to third-degree heart block. The approach to a VSD repair depends on its specific location within the ventricular septum. Defects under the pulmonary valve can be approached through an incision in the right ventricular outflow tract. Muscular VSDs, although more likely to close on their own, can be more difficult to repair surgically due to their location within trabeculations of the heart. Surgical closure can often require a ventricular incision. Many muscular defects are now closed with an endovascular device either through a percutaneous or periventricular approach. The most commonly surgically closed VSD is a perimembranous defect and is approached through the right atrium.
 < div class='tao-gold-member'>
< div class='tao-gold-member'>




Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
