Congenital Anomalies of the Esophagus
David H. Rothstein
Marleta Reynolds
Multiple theories have been proposed to explain the pathogenesis of esophageal anomalies. The classic theory, as presented by Gray and Skandalakis,21 describes the ingrowth of mesodermal ridges to divide the foregut into trachea and esophagus. Error in fusion of these ridges results in various forms of tracheo- esophageal fistulas (TEFs) (Fig. 147-1). O’Rahilly and Muller52 proposed that a TEF develops when an abnormal epithelium-lined connection forms between the developing esophagus and trachea. Kluth and colleagues39 have presented convincing data supporting the theory that the trachea and esophagus develop and divide from cranial, ventral, and dorsal folds of the foregut. The ventral fold divides the foregut into trachea and esophagus. Esophageal atresia with distal fistula results from excessive ingrowth of the dorsal fold and the formation of a proximal pouch. The incomplete fusion of the cranial and ventral folds results in a fistula. Isolated esophageal atresia may result from the interruption of the blood supply to the esophagus, as seen in other forms of gastrointestinal atresia.
Work in rodent models has suggested that esophageal atresia is secondary to vascular compromise of the developing foregut.11,16 In response, the lung bud develops a trifurcation instead of a bifurcation, with the middle branch descending to join the stomach. This hypothesis can be used to explain the poor motility of the distal esophagus and the presence of cartilage in some distal esophageal stenoses.
Esophageal Atresia and Tracheoesophageal Fistula
Incidence
Esophageal atresia occurs in 1 in 2,000 to 1 in 4,500 births in the United States, with a male-to-female ratio of 1:26, an increased risk with the first pregnancy, and a slight increase in rate with rising maternal age. There is also an increase in twin births and a 6% incidence of chromosomal anomalies in affected infants.22,23
Associated cardiac, genitourinary, gastrointestinal, and neurologic anomalies occur in 50% to 70% of infants with esophageal atresia. Tracheobronchial anomalies were reported by Usui and coworkers72 to occur in 47% of infants with esophageal atresia. Choudhury and colleagues10 found associated cardiac anomalies to reduce survival with esophageal atresia from 88% to 59%. Quan and Smith54 coined the acronym VATER to describe infants with esophageal atresia and TEF (EA/TEF), vertebral anomalies, imperforate anus, and renal and radial arm anomalies. Associated cardiac and limb anomalies expand the acronym to VACTERL. Although cardiac anomalies once contributed to a mortality rate of up to 24% in TEF patients with the VACTERL association, continual improvements in cardiac and neonatal care have decreased the mortality to approximately 6%.36
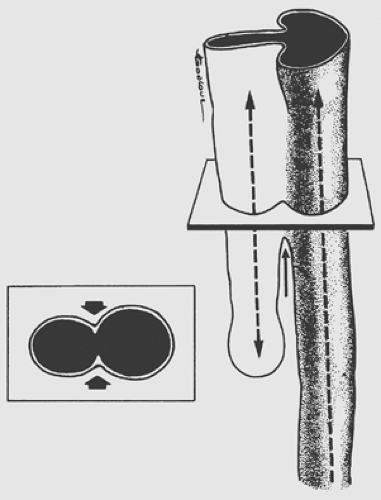 Figure 147-1. Division of the primitive foregut, with stippled area showing the future esophageal portion. Arrows indicate the local morphogenic movements. (From Gray SW, Skandalakis JE. Embryology for Surgeons: The Embryological Basis for the Treatment of Congenital Defects. Philadelphia: Saunders, 1972:64. With permission.) |
Several classification schemes have been used to describe the variants of esophageal atresia. Holder and colleagues26 reported the incidence of the most common variants of esophageal atresia (Fig. 147-2). Kluth38 collected every reported variation and created an atlas of esophageal atresia. Because of the number of variations, it is often easiest to describe the findings. Waterston and associates75 used risk factors to classify babies with esophageal atresia and recommend treatment. The risk factors included birth weight, pneumonia, and associated congenital anomalies (Table 147-1). Patients in group A, or the good-risk category, underwent immediate repair. Those in group B (the moderate-risk group) had delayed repair. The high-risk infants in group C were managed with staged repair. Improvements in the care of small babies have made this classification obsolete. Spitz and associates65 reported that birth weight and major cardiac anomalies were predictors of survival and grouped babies with esophageal atresia based on these factors (Table 147-2). Babies in group I undergo immediate repair. Most in group II also undergo immediate repair unless a palliative cardiac procedure is emergently indicated. With the use of prostaglandin, this is seldom necessary. Even now, babies in group III can undergo immediate or delayed repair, and a staged repair is almost never indicated.
Clinical Presentation
The diagnosis of esophageal atresia can be made in the fetus by ultrasound examination. The findings of an absent or small stomach bubble in conjunction with maternal polyhydramnios is highly suggestive of esophageal atresia. Vijayaraghavan74 also noted that the presence of a fluid-filled pouch in the neck of the fetus helps to confirm the diagnosis. Stringer and associates68 found the sensitivity of prenatal ultrasound in making the
diagnosis of esophageal atresia to be only 42%, with a positive predictive value of 56%.
diagnosis of esophageal atresia to be only 42%, with a positive predictive value of 56%.
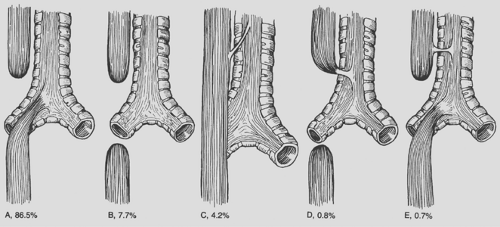 Figure 147-2. A: Esophageal atresia with distal tracheoesophageal fistula. B: Esophageal atresia without tracheoesophageal fistula. C: Tracheoesophageal fistula without esophageal atresia. D: Esophageal atresia with proximal tracheoesophageal fistula. E: Esophageal atresia with both proximal and distal tracheoesophageal fistulas. |
Table 147-1 Waterston Risk Classification of Esophageal Atresia and Tracheoesophageal Fistula | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Most babies with esophageal atresia present shortly after birth with excessive drooling. The astute neonatal nurse observes difficulty with the first feeding, choking, coughing, and even cyanosis. Early diagnosis can prevent the pulmonary complications that otherwise develop quickly. Aspiration of oral secretions from the proximal pouch can be prevented by passage of a tube, designed by Replogle,55 attached to continuous suction. If a distal fistula is present, the stomach quickly fills with air. Reflux of gastric secretions into the tracheobronchial tree may lead to chemical pneumonitis. As the abdomen becomes more distended, elevation of the diaphragm may further compromise the baby’s respiratory status.
Diagnosis
The diagnosis of esophageal atresia is made when a catheter placed in the proximal esophagus cannot be advanced into the stomach. A plain radiograph of the chest confirms the position of the catheter in the upper mediastinum near T1 or T2. Air can be injected through the catheter into the proximal pouch to better outline the pouch. Contrast studies used to identify a proximal fistula should be avoided owing to the risk of aspiration; a careful exam of the proximal pouch during repair can localize this rare anomaly.
A “babygram,” which includes the neck, chest, and abdomen, is used to diagnose a distal fistula, because the stomach and intestines have air throughout. The radiograph is also used to identify any associated anomalies of the heart, intestines, and vertebral column. The location of the aortic arch is important to some when planning the operative approach. If it is difficult to see on the plain radiograph, high-kilovolt images (Fig. 147-3) or echocardiography (Fig. 147-4) can be used. Canty and colleagues9 demonstrated an association of long-gap esophageal atresia (>3 cm between proximal and distal esophagus) with aortic arch anomalies. They recommend computed tomography (CT) or magnetic resonance imaging (MRI) if a right-sided arch is identified, because a vascular ring may need to be addressed at the time of esophageal repair and a left thoracotomy considered. The echocardiogram is also used to identify associated congenital heart disease.
Table 147-2 Survival Related to Birth Weight and Major Congenital Heart Disease, 1980–1994 | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
The diagnosis of an H-type TEF without esophageal atresia is often made late and with difficulty. A high index of suspicion and an experienced radiologist are needed. Water-soluble contrast or barium is slowly dripped into the proximal esophagus, and using fluoroscopic observation, the communication between the trachea and esophagus is identified in the lower neck and upper chest. Bronchoscopy with meticulous examination of the posterior wall of the proximal trachea can be used to make or confirm the diagnosis.
Esophageal Atresia with Distal Fistula
Initial Management
A thorough examination and evaluation of the baby with esophageal atresia with distal fistula should be completed before surgery. The initial babygram may identify duodenal atresia with the classic “double-bubble” sign. Imperforate anus can be ruled out with an inspection of the perineum and rectal examination. A cardiac echocardiogram and a renal ultrasound are excellent screening tools for associated anomalies.
In the past, timing of surgical correction was based on the Waterston classification (see Table 147-1). Improvements in perinatal care have made it possible to perform primary repair in smaller and smaller babies. Coexisting conditions, especially congenital heart disease and physiologic status, now dictate the timing and type of surgical repair. In the unstable baby, the proximal pouch is drained with the Replogle tube, and a gastrostomy tube and central line are placed (Fig. 147-5). The delayed repair is performed as soon as the baby’s condition permits. A staged repair, which consists of division of the fistula and delayed repair, is almost never indicated and would be reserved for babies too sick to tolerate repair but in whom ventilation is impossible without division of the fistula.
Operative Repair
Preoperative rigid bronchoscopy can be useful in identifying tracheomalacia and localizing the fistula site. Historically, a right posterior thoracotomy and extrapleural approach has been used to expose the esophagus and trachea. In the past several years, a thoracoscopic approach has been developed (see below). In the open approach, the pleura is reflected medially with blunt dissection, and the azygos vein is identified and divided. The distal esophagus and vagus nerves are immediately visualized. The fistula is usually located beneath the azygos vein, and the
distal esophagus enters the membranous trachea just above the carina. It can communicate with a major bronchus or higher in the trachea. The distal esophagus is isolated with a vessel loop, and the fistula is identified. The fistula is divided and the trachea closed with absorbable sutures. The tracheal closure is covered with pleura, the stump of the azygos vein, or other nearby tissue.
distal esophagus enters the membranous trachea just above the carina. It can communicate with a major bronchus or higher in the trachea. The distal esophagus is isolated with a vessel loop, and the fistula is identified. The fistula is divided and the trachea closed with absorbable sutures. The tracheal closure is covered with pleura, the stump of the azygos vein, or other nearby tissue.
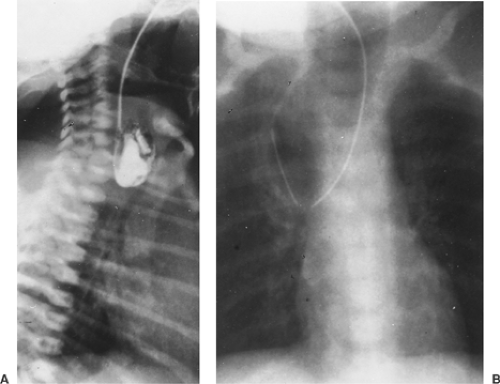 Figure 147-3. A: Esophagogram of an infant with esophageal atresia; barium outlines the proximal pouch. B: Radiograph of an infant with esophageal atresia. Air distends and outlines the proximal pouch. |
The proximal esophagus is located by downward pressure on the pouch applied with an esophageal stethoscope by the anesthesiologist. The proximal pouch is dissected free from adjacent tissue and incised at its tip. Davies12 cautions that extensive dissection of the proximal pouch leads to disruption of the nerve supply to the esophagus and may be responsible for subsequent problems with esophageal motility. Once the proximal pouch is opened, an undetected proximal fistula should be apparent if air bubbles into the proximal pouch from the trachea. If this occurs, a proximal fistula must also be divided from within the chest or the neck (Fig. 147-6). The small catheter is passed down the distal esophagus to ensure patency. The proximal and distal esophagus are approximated with a single-layer end-to-end anastomosis using absorbable suture. The anesthesiologist passes a small silicone polymer (Silastic) catheter through the nose and advances it through the anastomosis before the surgeon ties the
last suture. Some surgeons leave this catheter in place to provide a route for enteral feedings in the early postoperative period.
last suture. Some surgeons leave this catheter in place to provide a route for enteral feedings in the early postoperative period.
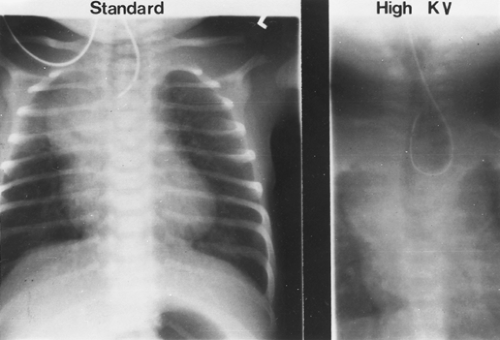 Figure 147-4. Standard and high-kilovolt radiograph of an infant with esophageal atresia. The high-kilovolt radiograph shows deviation of the trachea to the right, indicating a left aortic arch. (From McKneally MF, et al. Surgical treatment of congenital esophageal atresia. Ann Thorac Surg 1984;38:606. With permission.) |
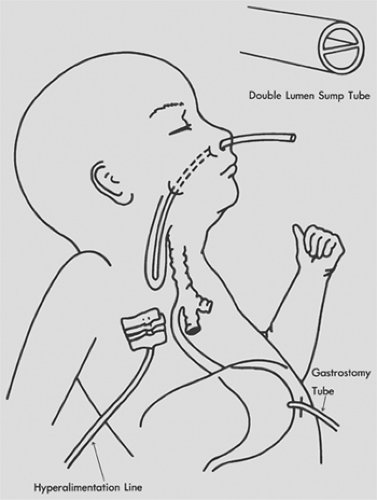 Figure 147-5. Replogle double-lumen sump catheter designed to decompress the proximal pouch in infants with esophageal atresia. (Adapted from Replogle RL. Esophageal atresia: plastic sump catheter for drainage of the proximal pouch. Surgery 1963;54:296. With permission.) |
Several techniques have been described to reduce the anastomotic tension in the case that the distance between proximal and distal esophagus is large (>2.5 cm). Single or multiple circular myotomies, as popularized by Livaditis and colleagues43 and de Lorimier and Harrison,15 can be performed to lengthen the proximal pouch (Fig. 147-7). Kimura and associates37 were proponents of a spiral myotomy to accomplish the same goal (Fig. 147-8). Either technique can be useful in the rare patient with a long gap between the two ends of the esophagus, but both have been implicated in worsening esophageal motility. More recently, Foker19 has described a technique of placing deep traction sutures on both ends of the esophagus and bringing the sutures out through the thoracic wall to apply continuous traction over the course of 7 to 10 days to promote esophageal lengthening. A repeat thoracotomy or thoracostomy is performed to create the final anastomosis without tension.
Before the chest is closed, a 10-Fr soft catheter with extra drainage holes is placed near the anastomosis and brought out of the chest through a separate incision. This is connected to water-seal drainage.
When the esophageal distance is too great for primary anastomosis, enteral interposition is performed. Techniques include
gastric pull-up, reverse gastric tube, and small- and large-bowel interposition.
gastric pull-up, reverse gastric tube, and small- and large-bowel interposition.
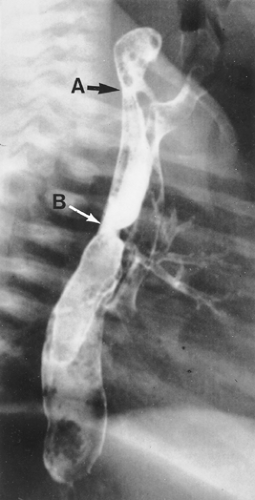 Figure 147-6. Esophagogram illustrating a high proximal fistula (A) that was missed at previous repair of esophageal atresia with distal tracheoesophageal fistula (B). |
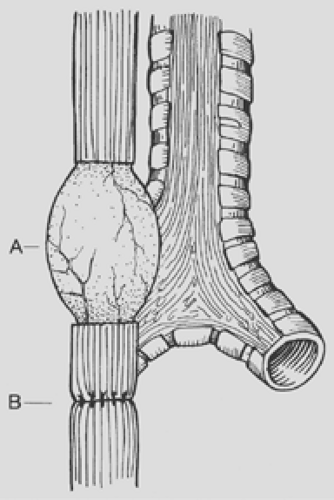 Figure 147-7. Esophageal myotomy (A) performed to increase the length of the proximal pouch in order to relieve tension on the esophageal anastomosis (B). |
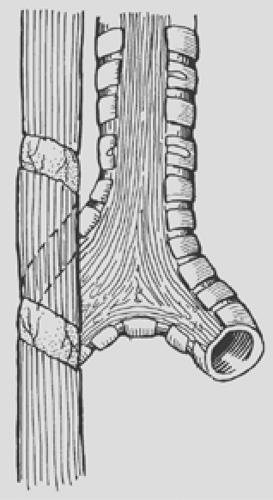 Figure 147-8. Spiral myotomy is an excellent technique for increasing the length of the proximal pouch in order to avoid excessive anastomotic tension. |
Thoracoscopic repair of esophageal atresia/tracheoeso- phageal fistula was first described by Bax and van der Zee5 as well as by Rothenberg59 in 2002. The procedure is performed in a transpleural approach through three or four 5-mm trocars under gentle carbon dioxide insufflation. The putative advantages are reduced operative trauma, decreased pain, and most importantly, a theoretical decrease in the likelihood of developing long-term abnormalities of vertebral and thoracic growth. Early results show no increase in short-term complications.25 A robotic approach has been championed by several groups but has not clearly been shown to have any advantages over the thoracoscopic approach.28
Postoperative Care
Attempts are made to extubate the baby in the operating room or as soon as possible in the newborn intensive care unit. To loosen secretions, these babies are kept in isolettes with high humidity. Deep suctioning is forbidden. Right-upper-lobe collapse occurs frequently but responds to gentle vibratory physiotherapy. Occasionally, laryngoscopy and direct aspiration of the trachea by the surgeon, who has direct knowledge of the location of the tracheal suture line, is helpful. If a nasogastric catheter has been left in place, feedings may be started within 24 to 48 hours of operation. Otherwise, oral feedings may be started after a barium swallow, obtained after 5 to 7 days, rules out an anastomotic leak (Fig. 147-9).
Nambirajan and associates48 have shown the barium swallow to have little value in the planning of subsequent care. They found that a radiologic leak did not alter management or outcome, and identification of an anastomotic stricture did not correlate with the need for dilation based on symptoms. The symptomatic stricture can be dilated after 2 to 3 weeks. Techniques for dilation include placement of a gastrostomy tube for retrograde dilation, blind prograde dilation, and balloon dilation under fluoroscopic guidance (Fig. 147-10). Because gastroesophageal reflux is present in virtually every infant with EA/TEF and can lead to anastomotic stricture, acid suppressive medications are routinely prescribed for the first 6 to 12 months postoperatively. Recent work by Uhlen and associates71 as well as others has suggested that topical application of the antiproliferative medication mitomycin-C during esophageal dilatation may reduce the incidence of recurrent stricture.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


