Prenatal ultrasound exam of a compressive CPAM of the left lung at 23 WG. The heart is displaced to the right by a macrocystic lesion of 58-mm diameter with compression of both lungs and secondary polyhydramnios but without hydrops. Prenatal thoracoamniotic shunting was decided.
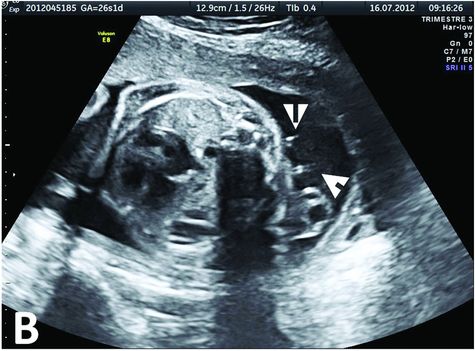
Prenatal ultrasound exam at 26 WG, 2 weeks after the CPAM drainage by two catheters (white arrows) placed in the predominant cysts. The cysts are well subsided, and the mediastinal shift is corrected. The drainage will remain functional until birth.
Prenatal treatment of hyperechogenic lesions is still debated. Sclerosis of the mass or coagulation of vascular pedicles has been proposed by some teams with mixed results[17]. Similarly, interventions by open surgery after hysterotomy and maternal fetal thoracotomy were performed for solid malformations. Series from Scott Adzick’s team are the largest to date, with 24 cases of in utero fetal thoracotomy within 15 years, carried out between 21 and 34 weeks. The fetal survival rate was about 50%. Half the deaths occurred intraoperatively during resection of the lesion and the other half in the hours following the intervention secondary to fetal bradycardia, uterine contractions or uncontrolled chorioamnionitis[26].
The decrease in prenatal volume of the malformations was estimated between 15 and 65%. This regression accounts for almost half of CPAM when the volume of the lesion is reported to total lung volume[26,27]. The ‘disappearance’ of congenital malformations of the lung on last prenatal ultrasound was classically described and interpreted as a complete regression of the malformation. This evolution, although exceptional, was documented for sequestrations[28] but not for cystic malformations, for which the ultrasound disappearance is attributed to changes in prenatal morphological feature at the third trimester. Thus, post-natal thoracic imaging (MRI or CT scan) is required to check the persistence of congenital lung malformations regardless of the prenatal evolution.
Post-natal diagnosis
Cystic lung malformations
Cystic lesions are congenital lung malformations for which complications are most often reported. In the neonatal period, respiratory distress secondary to compression of the lung parenchyma is the main symptom and leads to an emergency operation in the first month of life in about 20% of the children[21] (Figure 7.2).
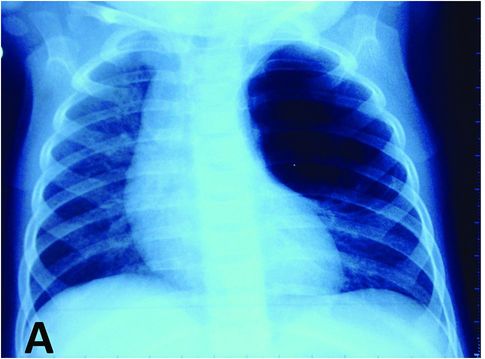
Macro-cystic malformation of the upper lobe of the left lung leading to post-natal respiratory distress. Chest X-ray shows cardiac shift with left mediastinal compression and tracheal deviation to the right.
On the other hand, bronchogenic cysts are small in the neonatal period and thus remain asymptomatic long after birth. Gradually, intracystic secretions lead to cystic growth and tensioning. This increase can be rapid and significant with symptoms related to the compression of adjacent structures: the trachea in most cases, more rarely the oesophagus or the vena cava and the myocardium. Cysts located just under the carena are particularly at risk and life threatening because of the simultaneous compression of both bronchi[29,30,31]. Although exceptional, symptoms can also be secondary to a fistula in the tracheobronchial tree, oesophagus or lung tissue[32,33].
After the neonatal period, infection is the most common complication of cystic malformations. It can be manifested by febrile episodes but also by recurrent respiratory symptoms that are usually diagnosed and treated as asthma[32]. Chronic inflammation may remain latent or be complicated by hemoptysis or pneumothorax[34,35]. In all theses cases, the diagnosis of the malformation, if undetected prenatally, or the confirmation of the complication is made on the chest CT scan (Figure 7.3).
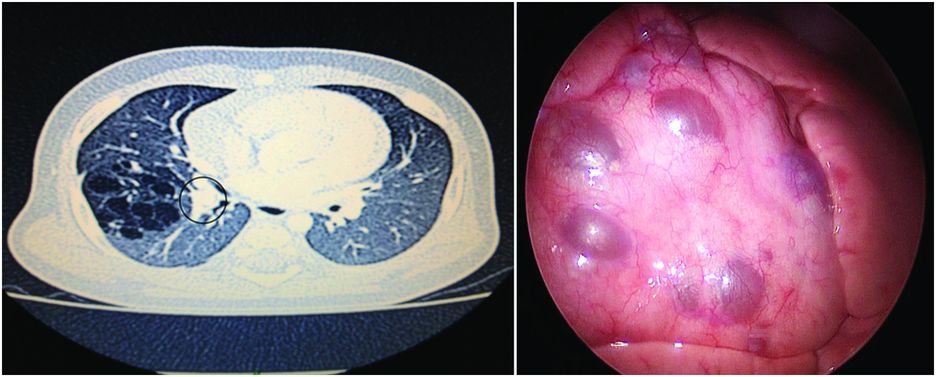
CPAM with multiple cysts: imaging and perioperative correlation.
Cystic malformation of the middle lobe of the right lung diagnosed after two episodes of pulmonary infection and abnormal chest X-ray. CT scan (left) shows numerous cysts with some thick dividing walls and proximal bronchocele (grey circle) leading to the diagnosis of associated bronchial atresia. Thoracoscopic per-operative view (right) shows the cysts at the surface of the middle lobe and the absence of lesions in the two other lobes.
To date, 50 cases of malignant degeneration of cystic malformations have been reported in children; the youngest age at diagnosis was 1 year[36,37]. Three kinds of tumours are described: pleuropneumoblastoma, bronchioloalveolar carcinoma and rhabdomyosarcoma. These tumours have been mainly described in CPAM but also, even though exceptional, in bronchogenic cysts[38,39]. In CPAM, the risk seems to depend on the type of cystic malformation: clearly associated to type 1 and type 4 CPAM when type 2 CPAM seems not involved[40,41]. Intracystic mucinous cell clusters, with K-ras mutations, have been described in type 1 CCAM, making these lesions potential precursors of bronchoalveolar carcinoma[42]. These concordant data strongly suggest that CPAM (at least type 1 CCAM) may predispose to malignant transformation. But the incidence of malignant transformation is still difficult to estimate: on systematic histological analyses, it was evaluated up to 4% in children’s CPAM and 20% in adults’ CPAM[43,44] and such differences may be reflecting the evolution with time of cystic congenital malformations[45,46].
Solid lung malformations
Solid malformation corresponds to bronchopulmonary sequestration (BPS). Two sub-types are described (i) intralobar sequestrations which share common visceral pleura with the normal lung and (ii) extralobar sequestrations which have their own pleura. Extralobar sequestrations account for 20% of prenatally detected BPS (Figure 7.4). Over 90% of sequestrations are found in the thorax, and less than 10% are located in the abdomen (all extralobar forms), typically described in the left suprarenal area. Because of this particular location, differential diagnoses such as neuroblastoma, adrenal hematoma, hemo-lymphangioma and teratoma have to be excluded[47]. BPS is characterized by a systemic vasculature that most commonly arises from the descending thoracic aorta, less frequently from the upper abdominal aorta (or celiac and splenic branches) and in rare cases from intercostal, subclavian or coronary arteries[48]. The existence of intradiaphragmatic locations is rare but is of embryological interest. The presence of abnormal tissue embedded within the diaphragm suggests that the sequestration forms prior to embryologic closure of the diaphragm, which is before the eighth week of gestation, and thus pleads for the origin from an early ectopic pulmonary bud. BPS with gastric duplications is also described[49]. The term is gastric duplication was proposed due to the fusion between the sequestration and the stomach, even in the absence of identified intestinal mucosa, considering that histological overlaps can occur because of the primitive digestive origin of lung tissues[50].
Bronchopulmonary sequestration: imaging and perioperative correlation.
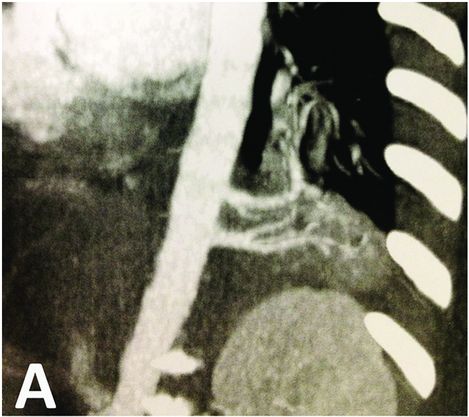
CT scan showing left basal bronchopulomonary sequestration with two feeding arteries arising from aorta. The malformation appears as a dense lesion without cystic component.
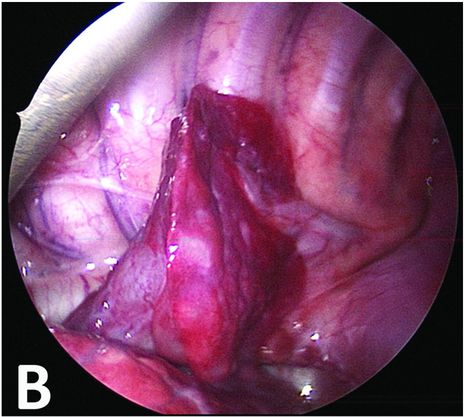
Thoracoscopic perioperative view confirming an extralobar sequestration with two feeding vessels.
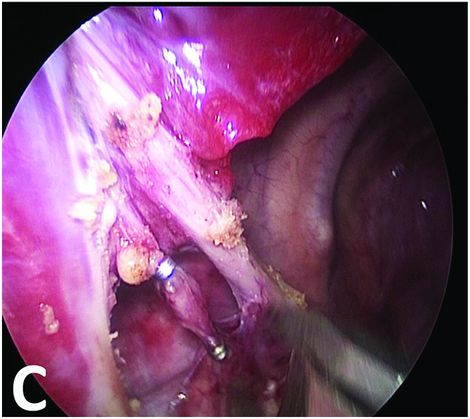
Dissection of the vessels and ligation by thoracoscopic clips.
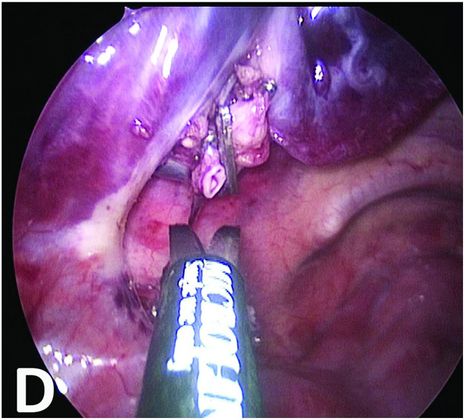
Section of the vessels and mobilization of the BPS.
In the post-natal period, symptoms are common to other malformations: respiratory discomfort and infection but more specifically, although exceptional, vascular complications, such as intralesional bleeding or heart failure[51,52]. Diagnosis appears earlier in extralobar sequestrations as 60% are recognized in the first 6 months of life, whereas intralobar sequestrations are most often diagnosed after the age of 2 years[50]. Sequestrations may also completely disappear in the pre- and post-natal periods, presumably by spontaneous thrombosis of the vascular pedicle, although histological data are not available to confirm the diagnosis. Expectant management can then be proposed for BPS, especially when the size of the lesion is small and if it is an extralobar form[34]. Percutaneous embolization could then be a good therapeutic alternative because of less morbidity, but it is reserved for BPS with a single artery and in the absence of associated cystic structures[53,54].
Emphysema
Emphysema is typically diagnosed in the neonatal period due to respiratory distress, acute or progressive effect by trapping and overinflation of the segment or the pulmonary lobe[55] (Figure 7.5). Although bronchus malformations were described at histological analysis in only half the cases, emphysema is attributed to abnormal bronchial cartilage without malformations of the lung parenchyma but secondary alveolar overdistension[56].
Congenital emphysema: imaging and perioperative correlation.
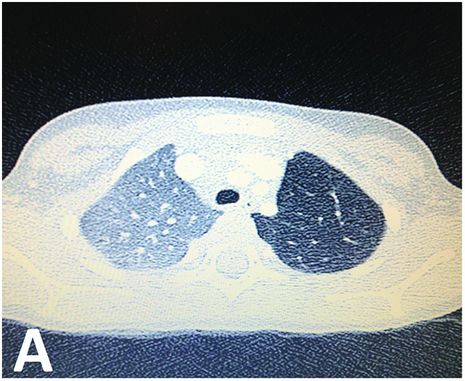
CT scan showing parenchymal hyperlucency of the upper left lobe.
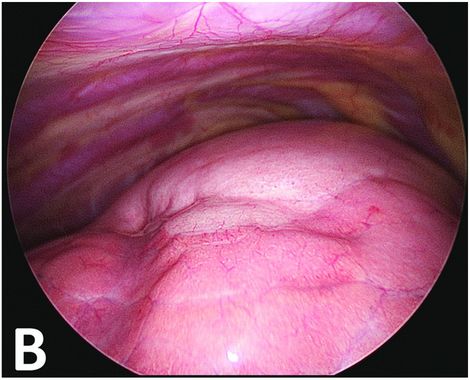
Thoracoscopic perioperative view. The upper part of the lung is discreetly clearer with very small superficial bubbles. The malformative zone can, however, be continuously delineated.
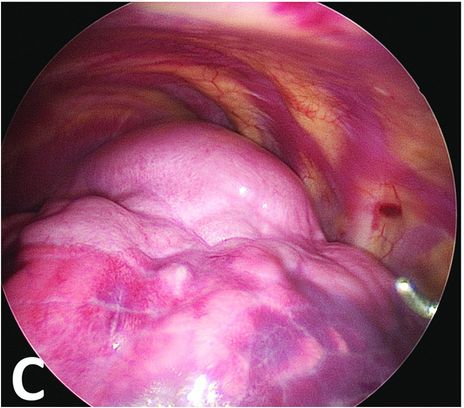
After ventilation exclusion of the left lung, the normal lower part is collapsed when the malformative upper part stays overinflated.
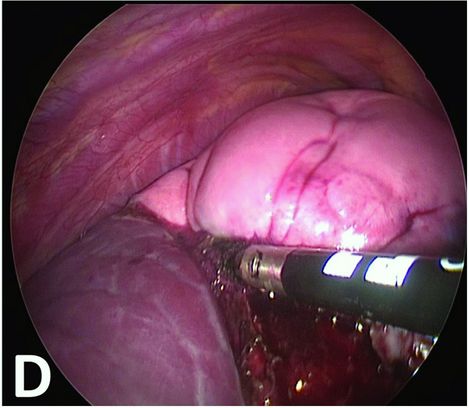
Thermal fusion parenchymal section at the edge of the emphysematous part.
Functional respiratory signs, with or without associated infection, are also common symptoms at diagnosis after the neonatal period. They occur in the majority of cases within the first 6 months of life[57]. In older children, it is sometimes difficult to tell the difference between late symptomatic congenital lobar emphysema and emphysematous lesions secondary to extrinsic bronchial compression or post-infectious lung damage[58,59].
Difficulty in breathing may also remain moderate with a favourable evolution and a regression of the lesions, provided there is not a local mechanical cause. Clinical improvements are associated to changes in ventilatory parameters[60].
Post-natal treatment
The principles of surgery in newborns are identical to those in older children, but the thoracoscopic approach, even if feasible as it was assessed by numerous publications, is difficult because of the limited space in the thorax at this age. As infants remain asymptomatic in the majority of cases, it allows an initial follow-up and avoids intervention and general anaesthesia in the first months of life; consensus seems to be emerging for intervention after 6 months[16,34].
At birth, chest X-ray is performed before discharge from hospital to assess the impact of the malformation once the lung is ventilated. CT scan or MRI, realized between 2 and 3 months of life, can precisely evaluate the morphological characteristics and the local impact of pulmonary malformation. The early term for this imaging, in the absence of planned surgery, has the advantage to still be performed at this age without general anaesthesia, which will not be the case in babies older than 3 months. In our experience, in the aim to limit irradiation, MRI provides a good pulmonary, mediastinal or vascular evaluation but fails to give a good evaluation of cystic components and can thus be completed by a low-dose scanner. This evaluation allows us to define precisely the type of malformation: strictly cystic (CPAM), entirely solid malformation (BPS) or cystic and solid components (i.e. hybrid malformation) in case of prenatal malformation with systemic pedicle; CPAM, BPS, emphysema or bronchial atresia in case of prenatal hyperechogenic malformations.
The surgical resection of lung congenital malformations is consensual when they become symptomatic: dyspnea, tachypnea, acute respiratory distress, discomfort in food intake, etc.[16,34]. For asymptomatic malformations (prenatally diagnosed), the main argument of the wait-and-see attitude is the possible disappearance of lesions over time. Disappearance was also documented for BPS[28,61], but there were no clear evidences of such evolution in cystic malformations.
The second argument for the expectative attitude is the poor knowledge of the natural history of congenital lung malformations, particularly with regard to delayed symptoms. Lobar emphysema may remain asymptomatic with little risk of major complications, and clinical and morphological improvements were even shown, justifying a long-term surveillance[60]. But even in the absence of functional symptoms, resection of cystic lesions is proposed. Although the evaluation of secondary infection is variable (from 10 to 85%), they were associated to higher perioperative complications with longer operativetimes, increased blood loss or higher rates of conversion when performed thoracoscopically[62]. Post-operative morbidity was also increased with longer lengths of stay and more frequent post-operative complications (pulmonary fistulae, secondary haemorrhage, reoperation, etc.)[14,63].
Early surgery is also recommended in cystic malformations because of the risk of malignant transformations[41,43]. Unlike bronchopulmonary carcinoma (cf supra), it is most likely that CPAM and pulmonary pleuroblastoma (PPB) are two separate lesions without natural evolution between them; chromosomal abnormalities that are identified in PPB cases are not found in CPAM cases[64,65]. Without complete resection, low-grade cystic PPB (type I) is likely to progress over 2–4 years to a high-grade solid disease (type III)[41]. Delayed recognition or failure to resect type I PPB significantly worsens prognosis. Overall survival rate in PPB ranges from 80–85% for type I to 45–50% for type III[66,67]. As these two entities share similar clinical and radiological features, pathologic examination is the only means to establish the final diagnosis[68]. Although some clinical features associated with cystic lung lesions can be helpful to suspect a type I PPB, they lack specificity and are often misinterpreted[66,67]. In their retrospective analysis of 51 patients with type I PPB, Hill et al. found that type I PPB was never suspected pre-operatively despite the high rate of pneumothorax at presentation and/or multiple lesions in the lung[67]. Thus, only systematic surgical removal of congenital cystic lung lesions, associated with a thorough pathologic study by a trained team, can formally exclude the diagnosis of PPB.
Cystic malformations (CPAM) with systemic vascularization or a predominantly solid lesion (BPS) but containing cystic structures, now defined as hybrid malformations, are thus to be considered for surgical resection.
Surgical procedure
The optimal period for lung resection seems to be between 6 months and 2 years. Surgery in the first 6 months for asymptomatic forms seems not necessary nor useful regarding the increased anaesthetic risk in the neonatal period compared to the incidence/consequences of complications. Moreover, the growth and development of the chest offer better technical conditions for thoracoscopic surgery after the first year. If resections are decided, interventions should be realized before the onset of complications, considering the data on increased morbidity, that is, in the first 3 years.
Progress in the miniaturization of equipment offers instruments of 3- or 5-mm diameters and 5-mm optics, perfectly suited for the thorax of infants. Lung resections, either thoraco-assisted or entirely thoracoscopic, are possible in the vast majority of cases, with an advantage (vs thoracotomy) on post-operative pain and costal healing[68]. In children, their low weight does not allow the use of a doublelumen endotracheal tube for transient pulmonary exclusion work, and the pulmonary exposure is more difficult than in adults. When the lesion is on the left lung, right selective intubation can be performed, if tolerated. When the lesion is on the right lung, a good collaboration with the anaesthesia team, providing a high-frequency and low-volume ventilation, is the key of this surgery.
The optical port is placed at the tip of the blade, on the mid-axillary line, and insufflation is performed at a maximum of 4–5 mmHg. Two 5-mm ports are positioned in triangulation location, and a third port may be necessary to improve the exposure. For parenchymal sections we used preferentially thermal tissue fusion (LigaSure⎩) that provides both control of the haemostasis and a good pneumostasis. Vascular controls, if necessary, are obtained by clips, endoloops or only by thermal tissue fusion, depending on the size of the pedicles. In children, the bronchi are flexible, and endoloop ligation is often sufficient without complementary stapling. In sequestrations, systemic arteries are controlled from the start of the intervention, venous return can be variable and there is no connection with the bronchial tree.
One or two chest tubes are left in place for an average of 48–72 hours. They are removed after ensuring the absence of bubbling and our usual test without prior clamping. Because of the lack of scholarship skin, a compression bandage is left for 48 h. The return home is made the day after the drains are removed after a final chest X-ray control.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


