Chronic Obstructive Pulmonary Disease: Epidemiology, Pathophysiology, Pathogenesis, and α1-Antitrypsin Deficiency
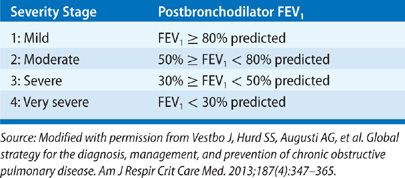
Chronic obstructive pulmonary disease (COPD) is defined as a disease state characterized by airflow obstruction that is not fully reversible and for which there is no other explanation for the obstruction. The obstruction is usually progressive and associated with an abnormal inflammatory response of the lungs to noxious particles and gas. Unlike earlier definitions, this definition does not mention emphysema or chronic bronchitis. According to criteria set by the Global Initiative for Obstructive Lung Disease,1 airflow obstruction is present when there is a reduction of the postbronchodilator FEV1/FVC ratio below 0.7 and its severity is graded by the percentage of the postbronchodilator FEV1 of the predicted normal FEV1 (Table 40-1). However, 0.7 as the cutoff value for the FEV1/FVC ratio has been controversial as it may be too high for all age groups and lead to misdiagnosis of COPD in healthy middle-aged and older individuals.2 Alternatively, use of the lower limit of normal (LLN) for the FEV1/FVC ratio has been recommended,3 but it is uncertain whether this criterion improves the detection of clinically significant COPD or prognosis in elderly individuals.4,5 Classification of COPD by GOLD has undergone further refinement recently with addition of self-reported severity of dyspnea and history of COPD exacerbations.1 Chapter 42 comments further about risk stratification.6
EPIDEMIOLOGY
COPD is a major health problem worldwide.7,8 Its prevalence is being recognized increasingly in countries at all levels of development.9 An ever-increasing number of smokers and an expanding number of elderly people are major factors in the surge in the worldwide prevalence of COPD. In a study from Canada, 27.6% of individuals reaching the age of 80 were diagnosed with COPD by a physician over the preceding 14 years.10 In large areas of the world where indoor air pollution is generated by burning biomass for heating and cooking, COPD is prevalent among nonsmokers, especially women.11 Moreover, COPD is not restricted to smokers in developed countries. Of 4291 never-smokers over age 40, involving 14 developed countries, 5.6% met criteria for moderate to severe COPD, of whom 81.2% were undiagnosed.12
At present, COPD is the third most common cause of death in the United States.13,14 As might be expected from the mortality figures, surveys indicate a high prevalence of COPD. The 2011 Behavioral Risk Factor Surveillance System (BRFSS), which is a state-based telephone survey of the US civilian adult population aged >18 years, found that 6.3% of US adults (an estimated 15 million nationwide) were told that they have COPD by a healthcare provider and a large percentage of these individuals reported having had spirometry.15 Another survey, the 2010 National Health Interview Survey of approximately 27,000 adults in US households, yielded an estimate of 5 million adults in the United States with emphysema and 10 million with chronic bronchitis, not all of whom may have airflow obstruction.16 Although these figures are impressive, they may be an underestimate, since COPD is likely to be unrecognized in some groups such as elderly persons living in poverty. Other statistics support these national surveys. Among 1575 cigarette smokers, aged 30 or older, with a 10 or more pack-year smoking history, approximately 20% met spirometric criteria for COPD.17
The number of deaths due to COPD in the United States has been rising. In 2008, COPD was the primary cause of death in 141,090 Americans.14 and was a comorbidity in many other deaths. The number of women dying of COPD now exceeds men and has more than doubled in recent decades. Based on the Third National Health and Nutrition Examination Survey (NHANES III), life expectancy is shortened by 5.8 years for men aged 65 with GOLD stage 4 COPD and by an additional 3.5 years if smoking has continued.18
ETIOLOGY
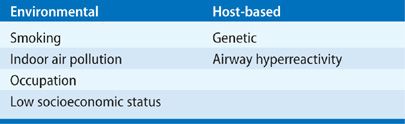
Risk factors for the development of COPD are environmental and host based (Table 40-2). In developed countries, smoking tobacco is the predominant risk factor. However, as noted above, never-smokers also develop COPD and women predominate in this cohort.11,12 In places where solid fuels are burned, indoor air pollution is probably the dominant risk factor. Other factors associated with COPD include second-hand tobacco exposure, age, level of education, tuberculosis, hospitalization for respiratory illness before the age of 10 years (see further comment below), a family history of COPD, and the number of years worked in dusty jobs.19 Clearly, multiple risk factors may be present in a single individual.
 ENVIRONMENTAL
ENVIRONMENTAL
Smoking
A history of smoking is present in 80% to 90% of the individuals with COPD in developed countries. In the United States an impressive level of overlap exists between the geographic distribution of smokers and the geographic distribution of individuals who have been told they have COPD (Fig. 40-1). In a European study involving 6836 individuals with normal spirometry, age 20 to 44, smoking was the most commonly recognized risk factor for the development of new COPD over a decade.20 Deterioration of FEV1 correlates with pack-years of smoking, but the relationship between amount of smoking and risk of COPD is unpredictable on an individual basis (Fig. 40-2), In the Million Women Study conducted in the United Kingdom, that involved 232,461 current, 328,417 former smokers, and 619,774 never-smokers at baseline, the relative risk of death from chronic lung disease, presumably mostly COPD, over a 12-year follow-up was increased about 35-fold among middle-aged women who smoked.21 Among former smokers, the age at smoking cessation affects the subsequent rate of deterioration of lung function. The rate is closest to never smokers for those who quit prior to age 30, but even for those who quit after age 40, deterioration is less than in continued smokers (Fig. 40-3).
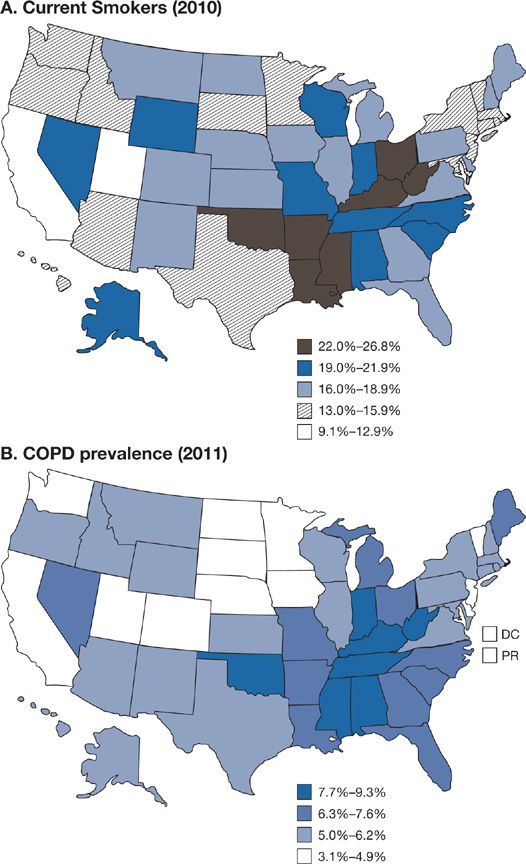
Figure 40-1 A. Percentage of persons aged ≥18 years who were current cigarette smokers, by state. Behavioral Risk Factor Surveillance System, United States, 2010. Persons who reported smoking at least 100 cigarettes during their lifetime and who, at the time of the survey, reported smoking cigarettes every day or some days. B. Age-adjusted prevalence of chronic obstructive pulmonary disease (COPD) among adults—Behavioral Risk Factor Surveillance System, United States, 2011. Based on an affirmative response to the question, “Has a doctor, nurse, or other health professional ever told you that you have COPD, emphysema, or chronic bronchitis?” (A. Reproduced with permission from Vital Signs. Current Cigarette Smoking Among Adults Aged ≥18 Years—United States, 2005–2010. Morbidity and Mortality Weekly Report. 2011;60(35):1207–1212. B. Reproduced with permission from Chronic Obstructive Pulmonary Disease Among Adults—United States, 2011. Morbidity and Mortality Weekly Report. 2012;61(46):938–943.)
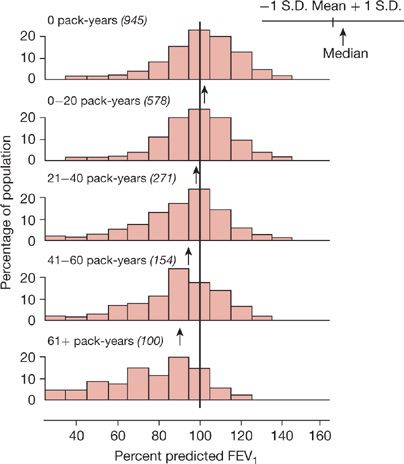
Figure 40-2 Distribution of percent predicted FEV1 in adults with varying pack-years of smoking. Subjects with “respiratory trouble” before age 16 are excluded. The proportion of smokers with normal expiratory airflow decreases with increasing pack-years. Nevertheless, many smokers have a normal FEV1 despite large cigarette-smoking histories. Means, medians, and ± standard deviation of the data for each group are shown in the abscissas. The numbers in parentheses are the numbers of subjects. (Reproduced with permission from Burrows B, Knudson RJ, Cline M, et al. Quantitative relationships between cigarette smoking and ventilatory function. Am Rev Respir Dis. 1977;115:195–205.)
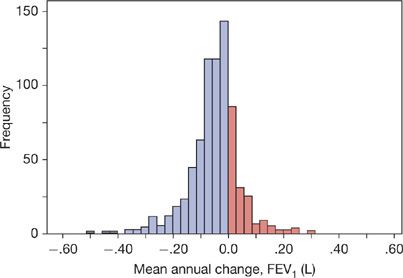
Figure 40-3 Histogram of the mean annual FEV1 decline (L) for 751 patients with COPD followed for a median of 64 months and up to 10 years, having an average of 5.44 annual measurements, average age 66 years, 92% male, stratified by ATS/ERS and GOLD severity classification as follows: 32 (4%) mild (FEV1% ≥80); 256 (34%) moderate (FEV1% 50–79); 245 (33%) severe (FEV1% 30–49); and 218 (29%) very severe (FEV1% <30). (Reproduced with permission from Casanova C, de Torres JP, Aguirre-Jaíme A, et al. The progression of chronic obstructive pulmonary disease is heterogeneous: the experience of the BODE cohort. Am J Respir Crit Care Med. 2011;184(9):1015–1021.)
The effects of gender on the risk of developing COPD from smoking are unclear. Women may be more susceptible to lung injury from smoking than men as they show more lung function reduction in association with lower total exposure and they predominate among individuals with early onset of COPD22 and never-smokers with COPD. However, among individuals with advanced COPD treated with long-term oxygen therapy, men have a higher mortality rate than women, a difference that cannot be explained by comorbidities.23 Some data indicate racial disparities in the risk of developing COPD, with African Americans showing similar severity of COPD to whites with lesser pack-years of smoking.24,25 Hispanic ethnicity appears to confer protection from the risk of COPD and a reduced risk of accelerated decline in lung function due to smoking.26
According to statistics from the Center for Disease Control and Prevention (CDC), in 2010 there were an estimated 45.3 million smokers in the United States representing 19.3% of all adults over age 18.27 The percentage was similar in African Americans and whites. Smoking was present in 21% of men and 17% of women. Hispanics and Asians had the lowest percentage of smokers at 12% and 9%, respectively, while the percentage was highest at 27% among American Indian/Alaska Natives. Beyond age 65 the incidence of smoking was 9.5%. The number of smokers worldwide is predicted to be 1.5 to 1.9 billion in 2025.28 Thus, COPD will continue to be a profound health problem well into the future.
Among smokers who have already sustained reductions in FEV1, the consequences of continued smoking on ventilatory function are more impressive than when all smokers are grouped together.29 The Lung Health Study, which followed middle-aged smokers for 11 years, found accelerated decline in those who already had a reduced FEV1 at the start.30 Despite these findings, progressive decline in the FEV1 is not inevitable among individuals with COPD (Fig. 40-4).31,32
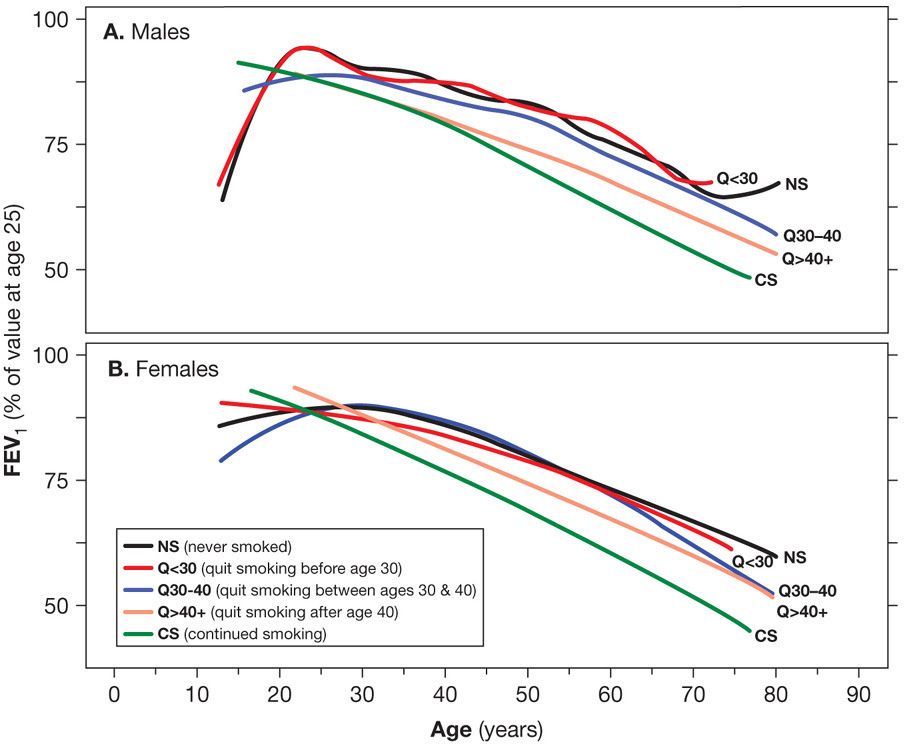
Figure 40-4 The effect of age of quitting smoking on FEV1. Mean FEV1 values (expressed as percent of its value at age 25) in smokers who quit smoking before the age 30 (Q < 30), between 30 and 40 years of age (Q30–40) and after age 40 (Q40+). Curves from healthy never smokers (NS) and continuous smokers (CS) are included for comparison. A. Males. B. Females. The mean annual FEV1 decline for males was 15.5 mL in quitters before age 30, 24.0 mL in quitters 30 to 40 years of age, and 28.9 mL in quitters after age 40; for females, the values were 10.4 mL quitting before age 30, 16.5 mL in quitters between 30 and 40 years of age, and 21.0 mL in quitters after the age of 40. (Reproduced with permission from Kohansal R, Martinez-Camblor P, Agustí A, et al. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10.)
Environmental Tobacco Smoke Exposure or Second-Hand Smoke
Environmental tobacco smoke exposure (ETS) is implicated in loss of many years of life of adults and children in the United States, with notable sensitivity among African Americans,33 but COPD, specifically, as a cause of the life shortening due to ETS is not clear34 Controlled experimental studies with normal volunteers indicate that short-term exposures to ETS at levels comparable to those in real-life situations have effects on serum cytokine levels and pulmonary function that if recurrent or chronic might translate into COPD.35,36 However, when smoking and other risk factors are controlled both workplace and home ETS but not prenatal ETS increase the risk of development of COPD.37 Data regarding in utero effects of maternal smoking on lung growth and subsequent risk of childhood wheezing or asthma are becoming evident. However, doubt exists regarding the quantitative impact on the development of COPD in individuals with only prenatal exposure. It seems likely that similar to cystic fibrosis, individuals with enhanced genetic risk factors could be adversely modulated by ETS,38 but to date no definitive proof of gene-by-environment interactions for ETS have been demonstrated. The data does not suggest that ETS is harmless but rather it is less definitively causal of COPD as an independent risk than chronic smoking or occupational exposures.37 Avoidance in individuals with existing lung disease is clearly indicated given the association with exacerbation. The attitude that there is no risk-free dose of ETS, is likely the safest approach; this applies to all ETS-related diseases and not just COPD.34
Indoor Air Pollution
As stated above, indoor air pollution from burning solid fuels such as wood and animal dung for cooking and heating is widespread in parts of the world. This practice exposes women and children to high concentrations of smoke containing respirable particles and complex gas mixtures for many hours daily and thus the risk of COPD. It compounds the risk of COPD in smokers who are more likely to be men in these settings. Cough and sputum occur among those exposed to indoor air pollution and an association with COPD is evident.39,40 Abatement of bronchitic symptoms coincident with measures that reduce the levels of smoke strongly implicates the indoor air pollution.41 The World Health Organization estimates that more than 1 million people a year die of COPD precipitated by indoor air pollution.42 International advocacy organizations such as the Global Alliance for Clean Cook-stoves seek to curb indoor air pollution.
Occupation
The possibility that occupation-related inhalation of particulates and gases carries a risk for COPD was slow in gaining acceptance, but acceptance is widespread now.43,44 The delay was understandable since smoking among workers in certain occupations was a confounding factor. Also, workers beginning jobs with a high risk of causing lung disease typically have better lung function than normal (the “healthy worker” phenomenon), obscuring work-related effects among relatively young workers. In addition, among cohorts of workers, those with COPD may drop out, causing an underestimate of risk in follow-up studies of individuals still working.
Despite these problems, studies from different groups around the world, urban and rural, workforce-based and community-based, clearly implicate occupations producing exposures to dusts, gases, and fumes as risk factors for COPD.44 The American Thoracic Society has estimated that the occupational contribution to the population burden of COPD is 15%.45 Apart from the well-recognized risk of occupations involving exposure to organic and inorganic dusts, less obviously “risky” occupations, such as construction, plastics manufacturing, and utility work may carry an increased risk of COPD.46 The risk of adverse occupational exposure is particularly important in workers who smoke or have other factors that raise their risk for COPD, such as α1-antitrypsin deficiency.47
Childhood Lower Respiratory Tract Infections
Since the status of lung function in very early childhood predicts ventilatory function many years later,48 it is plausible that lower respiratory tract infections (LRIs) during childhood might adversely affect lung development and increase the risk of developing COPD later in life. However, lung function in children who had pneumonia up to age 2 infrequently had reduced lung function 10 years after the infection.49 If there was a ventilatory defect, it was most often restrictive. Where reduced airflow was observed, an adenovirus was the predominant class of pathogens responsible for the pneumonia. It is notable that COPD exacerbations may leave only a minor lasting effect on airflow. Continued smokers enrolled in the Lung Health Study had only an additional loss of 7 mL of FEV1 per year for those having one exacerbation per year, while among those who had quit smoking, exacerbations had no permanent effect on the FEV1.50
 LOW SOCIOECONOMIC STATUS
LOW SOCIOECONOMIC STATUS
A low socioeconomic status is a risk factor for COPD. The relationship may be linked to an increased amount of smoking and other factors, including deficient medical care for respiratory infections, occupational exposure to inhaled particulates, and increased exposure to household allergens. The importance of smoking is evident. In the United States in 2011, 45% of adults with a General Education Diploma (GED) were smokers compared to 10% of individuals with a college degree, and among adults living below the poverty level, 33% were smokers in contrast to 20% of individuals living above the poverty level.51
 HUMAN IMMUNODEFICIENCY VIRUS INFECTION
HUMAN IMMUNODEFICIENCY VIRUS INFECTION
Individuals with human immunodeficiency virus (HIV) who smoke have an increased risk of COPD or more specifically emphysema development.52–54 The risk appears to be modulated by activation of alveolar macrophages with evidence of enhanced production of matrix metalloproteinases (MMPs) in these individuals.55 Although HIV can infect macrophages,56 it is unclear if this is a direct alteration due to HIV infection or a response to some downstream alteration of the suppression of innate immune responses, like chronic Pneumocystis infection.57 The occurrence of COPD and pulmonary hypertension in smokers with HIV appears to be more common in individuals with a high viral load and lower CD4 cell counts and not an adverse consequence of antiretroviral therapy.58 However, emphysema is not reversible and relation to viral load or recovery of CD4 cell counts is not straightforward. Other health issues like malnutrition may contribute. A multicenter study about the lung in HIV supported by the National Insitutes of Health is underway.
 HOST BASED
HOST BASED
Genetics
The field of COPD genetics is moving rapidly so that capturing this subject is aiming at a moving target. Aggregation of COPD in families and concordance of pulmonary function in twin studies have established a role for genetic predisposition to COPD.59–61 Moreover, the occurrence of reduced maximal expiratory airflow among nonsmoking first-degree relatives of individuals with early-onset COPD provides further support.62 Perhaps most compelling is the marked variability in development of COPD among smokers. Dissecting specific genetic factors that increase the risk of COPD has proven difficult. α1-AT deficiency illustrates this difficulty,63 where even among individuals with an identified genetic risk factor, wide, unexplained variability in the occurrence of COPD exists.64
However, deficiency of functional α1-AT represents the best-known genetic risk for COPD (see below). In this example genetic mutations discovered in small initial sample sizes can be replicated in larger analysis despite a significant gene-by-environment interaction. To this end, very rare mutations in the coding sequence of elastin cause cutus laxa and can also lead to COPD.65–67 Although Marfan and Ehler–Danlos syndromes cause lung parenchymal blebs68 and in mouse models demonstrate developmental of airspace enlargement,69,70 no human cohort has demonstrated clear penetration of a fixed obstructive lung disease phenotype.71
Polymorphisms of genes involved in protease–antiprotease balance, antioxidant function, inflammation, and immune responses have been implicated in COPD in studies utilizing candidate gene approaches (Table 40-3).72–74 However, none of these polymorphisms are consistently confirmed in other cohorts or large genome-wide association studies of lung function or COPD (Table 40-4).75–78 Failure of confirmation may indicate flaws in previous studies, but differences in COPD phenotypes, ethnic background, or other factors might explain discrepancies between previous studies and subsequent data. Despite misgivings of reproducibility in candidate gene studies it should be noted a minor allele frequency (MAF) of less than 5% typically excludes rare polymorphisms from GWAS style studies. Rare alleles may be quite important if the effect is large as demonstrated by α1-AT (SerpinA1) (Table 40-3).
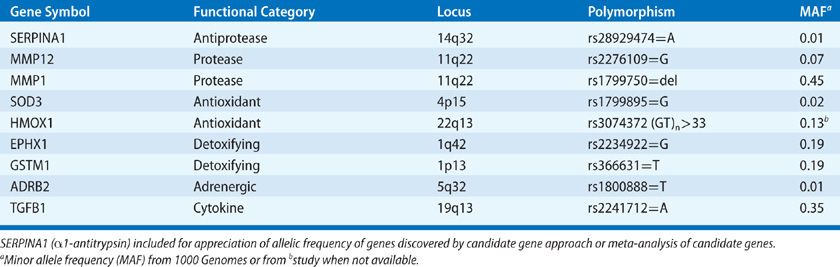
TABLE 40-4 Examples of COPD-Related Genes from Genome Wide Association Studies of Spirometry (prebronchodilator)
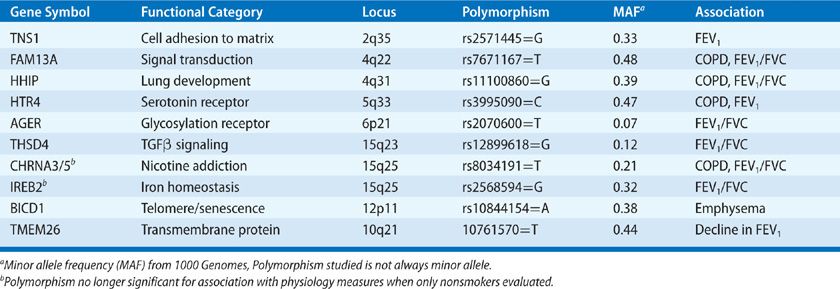
Traits like emphysema quantified by CT lung density, subjective dyspnea, and 6-minute walk distance can also be utilized as each trait has been independently associated with prognosis in COPD. Of note, analysis based on radiographic emphysema characterization does not seem to reveal the same genetic locations as airway physiology–based phenotyping (Table 40-4),79 possibly reflecting the advantage of extending genetic evaluation to other reproducible quantitative phenotypes, as differential genetic susceptibility to disease characteristics may reveal distinct independent pathways within the larger COPD population.
Very large GWAS studies of physiologic traits used to define COPD (FEV1, FEV1/FVC ratio) in populations that include nonsmokers and individuals too young to have COPD increase the understanding of how genetic factors interact with the environmental etiologic factors. For example, lung development and smoking habit may be teased out by examining COPD-susceptibility loci for association with the same traits in nonsmokers or younger individuals. This is important, as polymorphisms related to nicotine addiction80 (e.g., CHRNA3/5) need to be separated from polymorphisms that mark susceptibility to smoke exposure given the difficulty with quantifying lifetime smoke exposure. On the other hand, polymorphisms related to genes involved predominantly in lung development (e.g., HHIP) will result in alterations of the physiologic traits even in never-smokers, but may also augment increased susceptibility to smoking.81 It is notable that the different approaches of genetic analysis have not yielded as much cross-confirmation in COPD as has been seen in other diseases, despite the fairly large size of recent GWAS studies. In addition, although the same approaches have been utilized in asthma, a diagnosis that relies on the same physiologic traits, the polymorphisms discovered in asthma do not overlap COPD associated pathways.82–84 This distinct genetic framework of COPD versus asthma supports many years of work in mouse models defining divergent immune pathways in these two common airway diseases.85
Airway Hyperresponsiveness
Airway hyperresponsiveness (AHR) is present when there is an acute, temporary decline in maximal expiratory airflow in response to inhaling potential bronchoconstricting agents such as methacholine or histamine. In individuals with COPD, AHR is associated with accelerated decline of FEV1 and therefore is a negative prognostic marker.86 In the Lung Health Study, AHR was second to smoking as an important determinant of decline in FEV1 and was not related to the initial level of obstruction. AHR was greater in women smokers than men smokers.86 Although AHR decreased after smoking cessation and bronchodilator responsiveness improved, bronchodilator responsiveness did not correlate with the rate of FEV1 decline.87
A major question about the relationship of AHR to COPD involves consideration of the so-called “Dutch hypothesis,” which ascribes a role of allergy to the development of COPD. Observations that argue against AHR as a cause of COPD are that smokers typically do not show AHR until their FEV1 is already reduced and that experimental induction of emphysema can lead to AHR.88 It is plausible that AHR may be a consequence of emphysema, as airway wall stiffness and parenchymal tethering are affected by the extracellular matrix.89 If unmeasured emphysema is also connected to AHR, this may contribute to the gender bias and decline rate findings as emphysema presence may predict accelerated future decline regardless of smoking habit.90 As noted, the genetic distinction of asthma and COPD polymorphisms would also favor a great proportion of COPD having a nonasthmatic origin.
PATHOPHYSIOLOGY
A persistent reduction in FEV1/FVC is the defining physiological feature of COPD. Increased airway resistance, increased residual volume (RV), increased RV/total lung capacity ratio (RV/TLC), decreased inspiratory capacity, decreased maximum voluntary ventilation (MVV), abnormal distribution of ventilation, and ventilation–perfusion mismatching are also typical physiological features.
 AIRFLOW OBSTRUCTION
AIRFLOW OBSTRUCTION
The low FEV1 and low FEV1/FVC in COPD are not reversible with inhaled bronchodilators, although small improvements are common, especially if responsiveness is tested with both ipratropium and albuterol.91 Thus, COPD differs from asthma, in which inhaled bronchodilators can produce large improvements in FEV1. Maximal inspiratory flow may be relatively well preserved in the presence of a low FEV1/FVC and a low FEV1. A reduced FEV1 with a normal FEV1/FVC and normal TLC should not be interpreted as an obstructive ventilatory defect. This pulmonary function pattern is called nonspecific and infrequently progresses to COPD.92
The FEV1 is the result of the balance between the elastic recoil of the lungs promoting expiratory flow and the resistance of the airways that limits flow during performance of an FVC. In normal lungs, as well as in lungs affected by COPD, maximal expiratory flow diminishes as the lungs empty because the lung parenchyma provides progressively less elastic recoil and the cross-sectional area of the airways falls leading to an increase in airway resistance. The decrease in flow coincident with the decrease in lung volume is apparent on the expiratory limb of the flow–volume curve (Fig. 40-5). In mild COPD the abnormality in airflow is evident only at lung volumes at or below functional residual capacity, appearing as a “scooped out” lower part of the descending limb of the flow–volume curve. In advanced COPD, the entire curve demonstrates decreased expiratory flow. By measuring pressure–volume and pressure–flow relationships it is theoretically possible to assess the relative importance of decreased elastic recoil (“emphysema”) from increased airway resistance (“small airway disease”) as the cause for the reduced FEV1 As discussed below, the correlation between FEV1 and small airway pathology is strong and likely contributes to the reduction independent of the correlation of FEV1 with emphysema.
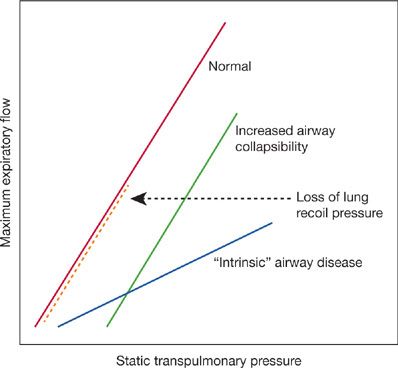
Figure 40-5 Analysis of reduced maximum expiratory flow in COPD from maximum expiratory flow versus lung recoil pressure curves. With loss of lung recoil pressure – that is, “emphysema” (heavy interrupted line) – the slope of the flow–pressure curve remains normal, but the curve terminates at lower pressure than normal. With intrinsic airway obstruction – that is, “bronchitis” – the slope is reduced. Increased airway collapsibility, which may be a result of decreased elastic recoil, causes the curve to be displaced to the right. Commonly in COPD, the flow–pressure curve has premature termination and a decreased slope and is shifted rightward, indicating that decreased elastic recoil, increased airway resistance, and increased airway collapsibility are all involved in causing the reduced maximum expiratory flow. (From Pride NB, Milic-Emili J. Lung mechanics, in Calverley PMA, Pride NB [eds]. Chronic Obstructive Pulmonary Disease. London, Chapman & Hall; 1995.)
There is wide variability in COPD in the relationships between FEV1, exercise tolerance, and quality of life.93 Variability also extends to the relationship between the FEV1 and alveolar gas exchange. However, the PaO2 and oxygen saturation usually remain near normal until the FEV1 has decreased to about half of the predicted normal while elevation of the PaCO2 seldom occurs until the FEV1 is less than about one-fourth of the predicted.94 Thus, other causes of hypoxemia or an elevated PaCO2, such as the obesity hypoventilation syndrome, should be considered in patients with abnormal arterial blood gases and only mild to moderate COPD. Similarly, pulmonary hypertension and right ventricular failure do not occur unless COPD is severe and associated with chronic hypoxemia (PaO2 <55 mm Hg). Diastolic dysfunction is common in the general population, where COPD is prevalent, and should be considered when pulmonary hypertension is discrepant with COPD severity.
 ABNORMAL DISTRIBUTION OF VENTILATION AND VENTILATION–PERFUSION MISMATCHING
ABNORMAL DISTRIBUTION OF VENTILATION AND VENTILATION–PERFUSION MISMATCHING
Abnormal distribution of ventilation results from the heterogeneity of the pathologic process affecting airways and lung parenchyma. This heterogeneous ventilation results in ventilation–perfusion mismatching that is characteristic of COPD. Abnormality in the distribution of ventilation is evident in the pattern of nitrogen washout during breathing of 100% oxygen. The nitrogen washout is delayed because of regions that are poorly ventilated, and the shape of the nitrogen washout curve reflects compartments with different washout rates due to regional differences in compliance and airway resistance. Radioisotopic ventilation scanning with 133xenon also reveals regional heterogeneity of ventilation in COPD, but can also demonstrate the ability of airway mucus to trap xenon tracer.
The multiple inert gas elimination technique (MIGET), which enables quantification of the ventilation–perfusion profile, has demonstrated different ventilation–perfusion patterns among patients with advanced COPD (Fig. 40-6). In one pattern, the so-called type A (“pink puffer”) COPD, there is a substantial amount of ventilation distributed to high ventilation–perfusion regions. This likely reflects actual loss of vascular elements in conjunction with the pathologic changes of the gas exchange parenchyma. In a second pattern, called type B (“blue bloater”) COPD, there is a substantial amount of pulmonary blood flow perfusing low ventilation regions. This simple classification was important historically in the development of understanding COPD pathophysiology, but has fallen out of use because most people with COPD are not easily classified as either type A or type B. They have both high and low ventilation–perfusion regions.
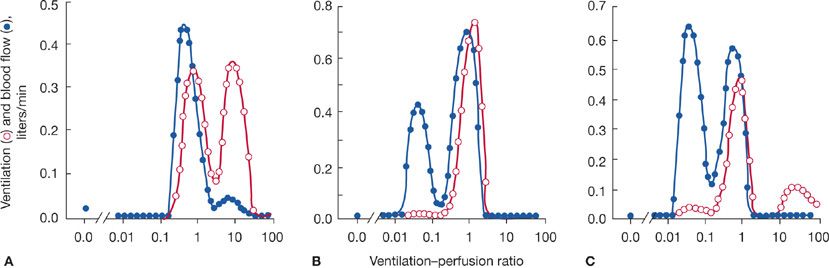
Figure 40-6 Ventilation–perfusion distributions in three persons with COPD determined by the multiple inert gas elimination technique (MIGET). A. Regions of high ventilation–perfusion characteristic of “emphysematous,” type A COPD. B. Regions of low ventilation–perfusion characteristic of “chronic bronchitis,” type B COPD. C. Regions of both high and low ventilation–perfusion characteristic of many people with COPD. In the normal person, not shown, ventilation–perfusion virtually overlaps and peaks at about a ventilation–perfusion ratio of 1. (Reproduced with permission from Wagner PD, Dantzker DR, Dueck R, et al. Ventilation-perfusion inequality in chronic obstructive pulmonary disease. J Clin Invest. 1977;59(2):203–216.)
Ventilation–perfusion mismatching accounts for essentially all of the reduction in PaO2 that occurs in COPD, so modest elevations of the inspired oxygen concentration are effective in treating hypoxemia. If hypoxemia is difficult to correct, other problems such as right- to left-sided intracardiac or intrapulmonary shunting need to be considered in addition to COPD.
 HYPERINFLATION
HYPERINFLATION
Increases in total lung capacity, functional residual capacity (FRC), residual volume, and the residual volume to total lung capacity ratio (RV/TLC) are common in COPD. These abnormalities may be beneficial in that they help to preserve expiratory airflow by increasing lung elastic recoil and the cross-sectional areas of airway lumens. However, they have adverse effects. They displace the diaphragm into a flattened position causing a number of adverse effects (Fig. 40-7). In addition, they put the thoracic cage at a mechanical disadvantage so that inspiration requires work rather than being passively assisted by the elastic recoil of the chest wall. These abnormalities of increased lung volume may increase further with exertion because reductions in airflow in diseased lungs reduce expiratory volume during rapid breathing. This phenomenon, called dynamic hyperinflation, adds to the workload on the inspiratory muscles while further reducing their mechanical advantage. Dynamic hyperinflation is an important mechanism of dyspnea with exertion in COPD.95
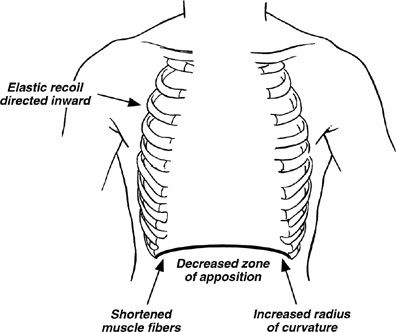
Figure 40-7 Detrimental effects of hyperinflation on diaphragmatic function. Hyperinflation causes flattening of the diaphragm, which (1) decreases the zone of apposition between the diaphragm and the abdominal wall, hindering rib cage movement; (2) shortens diaphragmatic muscle fiber length, decreasing the force that can be generated by the diaphragm; (3) increases the radius of curvature of the diaphragm, thereby decreasing transpulmonary pressure (at constant tension); and (4) directs diaphragmatic muscle fibers medially, impairing inflation with diaphragmatic contraction. In addition, hyperinflation prevents the thorax from assisting inspiration during tidal breathing because the resting volume of the thorax is above the volume at which the rib cage recoils outward during inspiration. (Reproduced with permission from Yusen RD, Lefrak SS. Evaluation of patients with emphysema for lung volume reduction surgery. Washington University Emphysema Surgery Group, Semin Thorac Cardiovasc Surg. 1996;8(1):83–93.)
Hyperinflation of the residual volume and FRC reduce the inspiratory capacity, which has prognostic significance that is independent of FEV1. In one study of individuals with moderate to severe COPD, those whose ratio of inspiratory capacity to total lung capacity was less than 25% had a much shorter life span than those with a ratio greater than 25%, even though the two groups had comparable percent predicted FEV1.96
 DYSPNEA
DYSPNEA
People with COPD typically seek medical care because shortness of breath limits their activities and quality of life. Dyspnea is seldom a complaint until the FEV1 has fallen below about 60% of predicted. However, the correlation between FEV1 and exercise limitation is not strong. Some individuals are relatively free of dyspnea despite a severely reduced FEV1. Commonly, the discomfort associated with breathing is associated with inspiration rather than expiration.97 Measurement of dyspnea has proven complicated.98 A number of indices are in use.
An increased sense of effort relating to the pressures needed from the respiratory muscles relative to their maximum pressure-generating capacity is thought to be an important factor in causing the dyspnea associated with COPD. Signals of “length-tension inappropriateness” from the respiratory muscles due to hyperinflation are another factor. Dynamic hyperinflation, as described above, exaggerates these problems for respiratory muscles. Also, impulses from airways undergoing abnormal dynamic compression during exhalation have been described. Hypoxemia and hypercapnia play only a small role during periods of clinical stability. Oxygen administration may decrease breathlessness by reducing ventilation during exertion and through poorly understood direct effects not associated with changes in ventilation.
PHYSIOLOGICAL–PATHOLOGICAL CORRELATIONS
In 1968, Hogg and colleagues observed that airways 2 mm or less in internal diameter normally contribute only a minor part of the total airway resistance, but that these airways are the principal sites of increased airway resistance in COPD.99 For many years, the physical basis for small airway resistance in COPD was considered to be a combined result of emphysema causing small airway instability and collapse along with multiple anatomic abnormalities narrowing the lumens of small airways. Because emphysema and small airway pathology are both common in individuals with COPD their relative contributions to airflow obstruction have been difficult to discern.
Fixed reduction in airflow is associated with several specific pathologic findings in the small airways of advanced COPD (Fig. 40-8). Small airways in the lungs of individuals with COPD typically show goblet cell metaplasia, replacement of Clara cells with mucus-secreting cells, and infiltration of the airway walls by inflammatory cells that, in severe disease, include an increased surface area of lymphoid follicles.100 The cellular changes are accompanied by increased connective tissue in the subepithelial and adventitial compartments of the airway walls.100 Alveolar tissue surrounding small airways normally provides radial traction on bronchioles at points where alveolar septa attach. Loss of these bronchiolar attachments as a result of proteolytic destruction may contribute to airway distortion, narrowing, and instability.
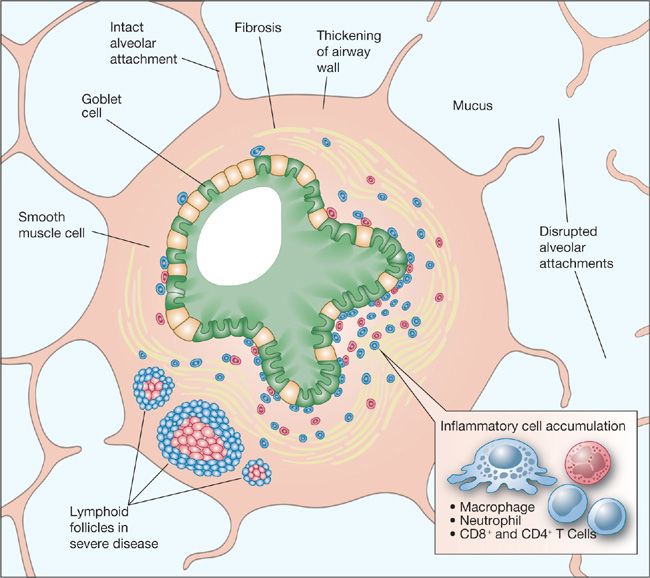
Figure 40-8 Pathologic lesions in small airways in COPD. Multiple abnormalities lead to partial obstruction of the lumen and altered shape and mechanical properties of the airways. (Reproduced with permission from Senior RM, Silverman EK. Chronic obstructive pulmonary disease. In: Nabel EG, ed. ACP Medicine: Pulmonary, Hamilton, Ontario. Canada: Decker Publishing; 2011.)
Although pathologic studies suggested the numeric loss of very small airways seen at autopsy is insufficient to produce the degree of airway resistance increases seen early in COPD, recent radiologic studies have confirmed there is a significant loss of visible (2–2.5-mm diameter) airways before radiographic emphysema is present.101 Indeed, the loss of small airways can be impressive, reaching 90% in severe COPD. The large-scale pathologic loss of terminal airways in advanced disease will contribute significantly to the small airway resistance101 but whether radiographic loss of small airway surface area in early disease is pathologic destruction, severe wall thickening or mucus plugging that obscures the lumen below the radiographic resolution is not yet clear. However, more severe airflow obstruction is clearly associated with increases in thickness of all components of the airway wall.100 The greatest relative increase in components of the airway wall is in the connective tissue–rich adventitial layer but the epithelial layer, the lamina propria, and the smooth muscle–containing layer all demonstrate significant increases in advanced disease and small airway mucus plugs have been demonstrated to be significantly associated with airflow limitation in severe disease. Thus, overall, compared to small airway pathology and radiologic loss of small airways, emphysema occurs late in the development of COPD and appears to have a limited role in causing the airflow obstruction in early COPD.102
PATHOGENETIC MECHANISMS
COPD represents the clinical expression of complex alterations in structure and function of alveolar tissue and small airways. Many processes at the tissue and cellular levels can be implicated, including inflammation, cell proliferation, apoptosis, altered phenotype of lung cells, and remodeling of the extracellular matrix (Fig. 40-9). Numerous mediators, most notably proteinases, oxidants, and cytokines, are involved in these processes. Studies in genetically altered mice have proven invaluable in helping to elucidate the pathogenesis of COPD, especially emphysema.
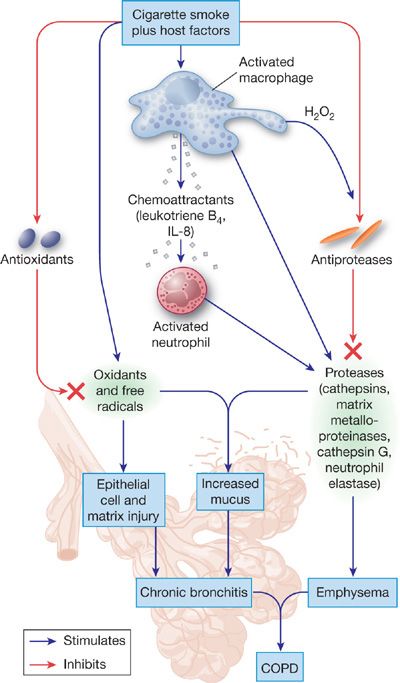
Figure 40-9 The pathogenesis of COPD from smoking. Smoking stimulates resident cells to release factors that recruit inflammatory cells to the lungs. The various inflammatory cells that accumulate in the peripheral tissues of the lungs release proteinases and oxidants that damage or degrade extracellular matrix in the walls of alveoli, alveolar ducts, and respiratory bronchioles. In addition, agents in smoke and those released by inflammatory cells inactivate proteinase inhibitors such as α1-antitrypsin, and cause senescence and apoptosis of lung cells that produce extracellular matrix. Products of the damaged extracellular matrix, such as peptides of degraded elastin, are chemotactic for inflammatory cells; thus degradation of the extracellular matrix may lead to a feedback loop that perpetuates inflammation. These matrix-derived products may also elicit immune responses that lead to destruction of extracellular matrix. Not shown are the role of mechanical forces that may also promote deformation of lung tissue. (Reproduced with permission from Senior RM, Silverman EK. Chronic obstructive pulmonary disease. In: Nabel EG, ed. ACP Medicine: Pulmonary, Hamilton, Ontario. Canada: Decker Publishing; 2011.)
 INFLAMMATION
INFLAMMATION
Innate Immune Responses
As reflected in the definition of COPD, inflammation occupies a central role in current thinking about the pathogenesis of COPD. The inflammation paradigm is that smoking and other types of inhaled irritants lead to recruitment of innate inflammatory cells to the lungs and airways and that products of these recruited cells injure lung tissue and disrupt normal mechanisms of lung repair. Indeed, inflammation is prominent in airways and lung parenchyma in biopsies, surgical specimens, and postmortem material from individuals with COPD.100,103–105 Other indicators of inflammation are increased inflammatory cells in bronchoalveolar lavage fluid (BALF)106–108 and sputum109–111 and increased volatile products of inflammatory cells in exhaled breath.112,113 Systemic inflammation is also present in current smokers, with elevations in white blood cell counts, neutrophil subsets, or liver-derived acute phase reactants.114–116 Inflammatory cells associated with COPD in the lung include predominantly neutrophils, macrophages, and sometimes eosinophils, but also dendritic cells and lymphocytes (see Acquired Immune Responses). Once the inflammatory process is initiated by smoking the process may persist long after smoking has stopped.117 Systemic neutrophil counts generally decrease within weeks but activated alveolar macrophages may be present even years after smoking cessation.117–119
Unlike nonsmokers, macrophage accumulations are found specifically in respiratory bronchioles, even in young smokers, and BALF from smokers contains many fold increases in macrophages compared to the numbers in BALF from nonsmokers.120,121 Besides releasing proteinases that might degrade the extracellular matrix of the lung,122 alveolar macrophages in COPD make chemotactic factors that recruit other inflammatory cells to the lungs. Likewise, structural cells of the lungs in COPD produce proteinases and chemotactic factors for inflammatory cells.123,124 Expression of interleukin-8 (IL-8), macrophage inflammatory protein-1α (MIP-1α), and monocyte chemoattractant protein-1 (MCP-1), for example, are upregulated in bronchiolar epithelium in COPD.125,126 Peptides of elastin are chemotactic for inflammatory cells and may act as epitopes for T-cell responses.127–129 In mice, genetically induced overexpression of cytokines, such as IL-13 or γ-interferon by lung cells leads to emphysema via a robust innate immune response, with inflammatory cell proteinases being integral in emphysema pathogenesis.130–132
Acquired Immune Responses
Cellular and humoral immunity may also be involved in emphysema pathogenesis or the continued progression after smoking cessation. CD4+ and CD8+ T cells and B cells accumulate in alveolar and airway tissue in COPD and form bronchus-associated lymphoid tissue (BALT) in the walls of small airways.100 The increasing BALT presence in small airways correlates with severity of GOLD stage.100 In mice, exposure to antibodies directed at endothelial cells alone elicits alveolar septal cell destruction and emphysema.133 Speculation about antigens for immunologically driven emphysema in patients include microbial pathogens, peptides altered by tobacco smoke, and peptides released from lung extracellular matrix.128,129,134 Difficulties in distinguishing cellular and humoral responses to microbial colonization of advanced airway disease in COPD from pathologic self-directed immune responses will require further study,135 but more targeted immunosuppresion in treating advanced COPD has not yet shown benefit.136,137 Intrinsic in this issue is the accelerated emphysema in smokers with HIV, but that may be complicated by direct virus infection inducing macrophage alterations, rather than suppression of acquired immune responses.55,56
 PROTEINASE–ANTIPROTEINASE IMBALANCE
PROTEINASE–ANTIPROTEINASE IMBALANCE
The discovery in the 1960s of α1-AT deficiency associated early-onset emphysema and the production of emphysema in experimental animal models with elastolytic enzymes have promoted the imbalance of proteinases relative to their inhibitors as a key factor in emphysema development.138,139 Although additional mechanisms, like apoptosis and oxidant stress, have been uncovered in recent years, the importance of proteinase excess continues to prevail as an important mechanism in emphysema development.
Proteinases of several biochemical classes, and different specific inhibitors, are implicated in the pathogenesis of emphysema. Serine proteinases, especially neutrophil elastase, and several matrix metalloproteinases, have been the proteinases for which there are the most data.140–143 It is notable that both neutrophils, which are the source of neutrophil elastase and MMP-12 from alveolar macrophages are largely related to continued smoking. Progression after smoking cessation may follow different pathways. As discussed in the genetics section many of these genes have been implicated in candidate gene studies but not genome-wide association studies (Tables 40-4 and 40-5).144–146 Although neutrophil elastase and its main inhibitor α1-AT have predominated the proteinase–antiproteinase imbalance hypothesis, MMPs appear prominent in mouse models and in samples from smokers and individuals with COPD. It is likely a combination of many local imbalances involving different proteinases and antiproteinases contribute to the progressive lung destruction.
Several aspects of proteinases in COPD should be noted, as a straightforward destructive mechanism only is likely an oversimplification. In addition to destruction of lung elastin and other matrix components, proteinases process cytokines and surface receptors involved in the inflammatory and immune responses.147–151 Inflammatory cells may not be the exclusive sources of the proteinases as structural cells also produce matrix-degrading proteinases.152 Even the apparently simple emphysema model of placing elastases in the lungs of experimental animals results in complex responses that can be altered by nonproteinase-related mechanisms including stem cell and immunologic responses.153,154 It must also be emphasized that little is known about proteinases in the pathogenesis of the small airway pathology of COPD. Virtually all of the information about proteinases in COPD pertains to emphysema pathogenesis despite clear evidence of small airway obliteration in advanced disease.
 OXIDANT–ANTIOXIDANT IMBALANCE
OXIDANT–ANTIOXIDANT IMBALANCE
Reactive oxygen species in cigarette smoke or released by inflammatory cells and structural cells of the lungs in response to smoke may lead to lung injury (see Chapter 41). Up to 20 mg of tar may be deposited in a smoker’s lung per cigarette smoked. This tar contains more than 1017 stable, long-lived radicals per gram. The gas phase of tobacco smoke contains 1015 organic radicals per puff of smoke, although in general these small oxygen- and carbon-centered species are more short-lived and reactive than the radicals in the particulate phase. In addition, tobacco smoke appears to “prime” neutrophils and alveolar macrophages to generate elevated amounts of reactive oxygen species, such as hydrogen peroxide, hydroxyl radicals, and superoxide radicals. The lung tissue of smokers contains significantly more iron than that of nonsmokers,155 providing a catalyst for the production of hydroxyl radicals from H2O2. This is of interest given the finding of an iron-binding protein polymorphism in the genome wide association studies of smokers with COPD, IREB2 (Table 40-4). Smokers also demonstrate increased production of neutrophil myeloperoxidase,106,112 which is capable of yielding oxidized halogens such as hypochlorous acid (HOCl). Oxidants modify and inactivate proteins, such as protease inhibitors (α1-AT and secretory leukoprotease inhibitor), and histone deacetylase 2 (HDAC2), which is involved in glucocorticoid mediated anti-inflammatory responses. Oxidants can affect lipids, DNA, and some specific end products, such as 4-hydroxy-2-nonenal (4-HNE) and 8-hydroxy-2′-deoxyguanosine (8-OHdG), may be markers of COPD.156,157
Oxidants can promote inflammation and proteinase expression, facilitate proteinase-mediated extracellular matrix degradation by enhancing matrix molecule susceptibility to proteolytic cleavage, and participate in nonenzymatic degradation of matrix molecules like type I collagen. In experimental animals the combination of cigarette smoke and elastase leads to greater emphysema than either insult alone, suggesting that these insults do not elicit identical responses.158 Animal models of antioxidant deficiency result in increased susceptibility to both cigarette smoke and direct elastase-induced disease.159–161
 APOPTOSIS AND SENESCENCE
APOPTOSIS AND SENESCENCE
Emphysematous human lung specimens demonstrate increased apoptotic and senescent cells compared to healthy lung specimens.162,163 An early theory of emphysema development was that alveolar vascular destruction preceded loss of alveolar tissue. Consistent with this early theory, the blockade of vascular endothelial growth factor (VEGF) signaling in alveolar endothelial cells or genetic downregulation of VEGF production in alveolar epithelium produces apoptosis and noninflammatory emphysema in rodents.164 In vitro, cigarette smoke induces apoptosis of several lung cell types.157,165,166 An important feature of experimental models of emphysema due to apoptosis is that there is minimal inflammation.167 Of interest, the BICD1 gene polymorphism linked to emphysema (Table 40-4) encodes for a protein in the apoptosis pathway.79 In contrast to the expanding body of information linking emphysema to apoptosis, there is only scant information about apoptosis of the cells of small airways in COPD.168 Much remains to be learned about apoptosis in the context of COPD airway disease.
Senescence of lung cells as a cause of emphysema stems from the knowledge of alveolar loss with aging and animal models169,170 where accelerated aging results in emphysematous changes. Lung fibroblasts isolated from human lungs with COPD demonstrate increased markers of senescence and senescent fibroblasts do not maintain the extracellular matrix.171 However, much of the information regarding telomeres in human COPD relates to inflammatory cell telomere shortening, with telomere length being a biomarker of chronic lifelong inflammatory excess present in individuals with COPD.172 Whether lung epithelial cells are driven to an injury-related replicative senescence is unknown, but human diseases of telomere deficiency and excess alveolar epithelial apoptosis tend to result in pulmonary fibrosis and not COPD.173,174
 MUCUS HYPERSECRETION
MUCUS HYPERSECRETION
Airway mucus is a normal protective barrier that is constantly replenished and cleared in health (see Chapter 6). Mucin glycoproteins, the main components of mucus, have a core protein rich in serine and threonine, to which carbohydrates and cysteine residues are attached. Mucus is secreted from submucosal glands and airway goblet cells. In COPD there is hyperplasia of goblet cells and hypertrophy of glands with an increase in the ratio of glandular mucus cells to serous cells. The changes in COPD are associated with an alteration of the mucus proteins (MUCs) to favor a predominance of MUC5B over the typical MUC5AC form, and an increase in the MUC2 form, which is uncommon in normal lung mucus.175,176 Other alterations in the mucus layer in COPD include greater acidity, less mucin glycosylation, and decreased antimicrobial peptides. Mediators responsible for mucus hypersecretion include proteinases, cytokines, oxidants, and epidermal growth factor receptor (EGFR) ligands.177,178 The negative charge of mucus glycoproteins results in sequestration of proteases, volatile hydrocarbons and possibly preservation of the hydration of the ciliated layer, resulting in protection of the underlying lung and likely improved carcinogen clearance. However, the symptoms of mucus hypersecretion are common complaints in individuals with COPD; quantity and location of mucus may be particularly important in symptomantic COPD.
Determining the relationship between chronic cough and sputum in patients with COPD and the natural history of COPD has been elusive.179 Reports vary from finding weak to strong correlations between cough and sputum production and COPD progression, COPD exacerbations, and mortality.180–182 A relationship between chronic mucus hypersecretion in small airways and adverse outcomes is plausible as histological analysis of small airway pathology in COPD demonstrated that the extent of small airway luminal obstruction by mucus correlated with the GOLD stage and was inversely correlated with survival after lung volume reduction surgery.104 Whether the mucus glycoproteins are a beneficial factor that mark the degree of inflammation (e.g., a biomarker of inflammation) or are themselves a pathologic factor in the severity of symptoms or progression of disease is an important question, as treatment of mucus hypersecretion without adequate suppression of the inciting inflammation may result in undesired consequences.182,183
PATHOGENESIS OF EMPHYSEMA
 GENERAL CONCEPTS
GENERAL CONCEPTS
Emphysema is defined as “a condition of the lung characterized by abnormal, permanent enlargement of airspaces distal to the terminal bronchiole, accompanied by destruction of their walls, and without obvious fibrosis.”184 However, studies since this definition was enunciated indicate increased collagen per unit volume of airspace wall in emphysematous tissue185 and active expression of elastin production (Fig. 40-10).186 Thus, emphysematous lung tissue should be viewed as actively undergoing remodeling rather than as inert. Further indications of activity in emphysematous lung tissue include the presence of many inflammatory cells103 and cellular changes of apoptosis and senescence.187
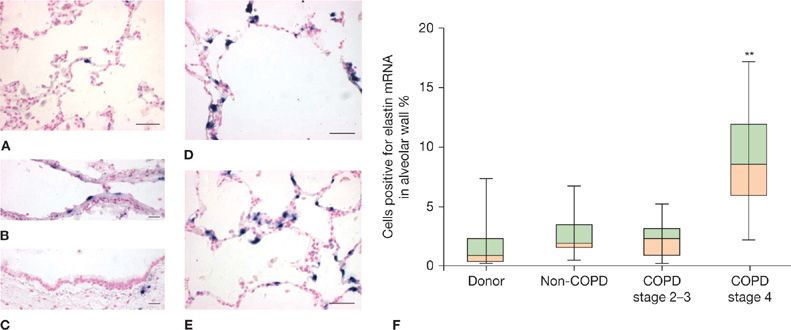
Figure 40-10 Active expression of elastin production in severely emphysematous lung removed for lung transplantation. Elastin mRNA (blue signal) as detected by in situ hybridization is rarely detected in donor lungs in (A) parenchyma, (B) intralobal pulmonary arteries or (C) airways, but is prevalent in parenchyma of GOLD 4 COPD lungs in regions with moderate (D) and severe (E) alveolar enlargement. Quantification of cells positive for elastin mRNA (F) reveals significantly higher elastin expression in alveolar walls of GOLD 4 COPD lungs compared to donor lungs or less severe COPD lungs. (Reproduced with permission Deslee G, Woods JC, Moore CM, et al. Elastin expression in very severe human COPD. Eur Respir J. 2009;34(2):324–331.)
Degradation of lung elastin by elastase activity from inflammatory cells is probably the predominant mechanism for emphysema in most smokers. However, the biology of emphysema is clearly complex and still incompletely understood. It includes inflammatory cell recruitment, proteinase–antiproteinase imbalance, oxidant–antioxidant imbalance, and responses of lung cells to proteinases and oxidants from inflammatory cells and to constituents of tobacco smoke. It may also involve humoral and cellular immunity. Degradation of extracellular matrix components besides elastin, particularly collagens, may be an important feature. In some situations, apoptosis of lung cells may precede degradation of extracellular matrix. Senescence of lung cells, a recently identified phenomenon in emphysema, has unclear implications, but suggests that lung repair mechanisms are depressed.
According to the proteinase–antiproteinase hypothesis, there is a constant or episodic release of proteinases, active at neutral pH, into the lung parenchyma. These proteinases come principally from inflammatory cells (Table 40-5). Under normal conditions circulating proteinase inhibitors, especially α1-AT, and inhibitors produced locally in the lungs, permeate lung tissue and prevent these proteinases from digesting the structural proteins of the lungs (Table 40-6). Emphysema results when the balance between proteinases and antiproteinases in lung tissue tilts in favor of proteinases due to increased proteinase release into the tissue and/or a reduction in the antiproteinase content in the tissue. The proteolytic events leading to emphysema occur in microenvironments adjacent to lung cells; proteinase–antiproteinase balance may not be reflected in measurements of large samples of emphysematous lung tissue.



