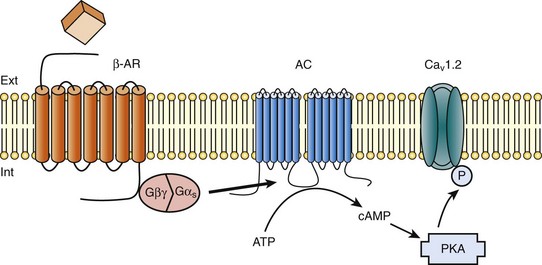
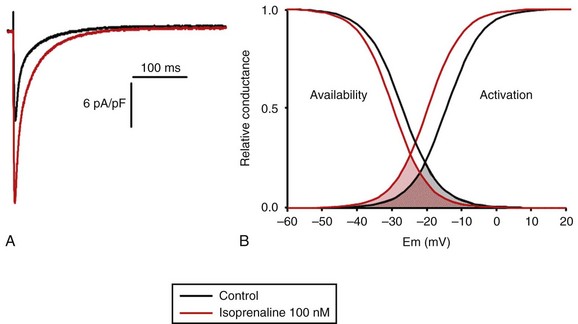
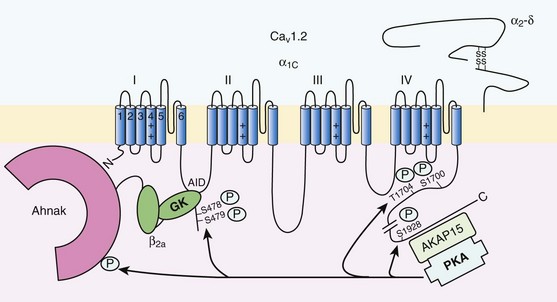
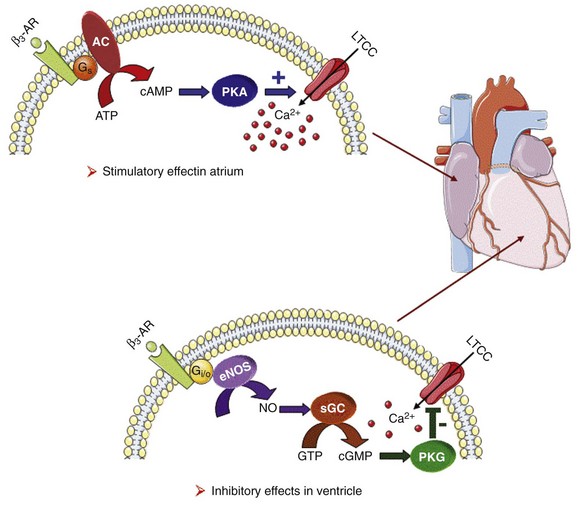
37 In cardiac cells, the L-type calcium channel (LTCC) current also called ICa,L underlies the plateau phase of the action potential (AP). Upon depolarization, ICa,L reflects calcium influx via the CaV1.2 channels. This current initiates cardiac contraction by gating the ryanodine receptor, thereby triggering the calcium release from the sarcoplasmic reticulum.1 Among several regulatory pathways of this current, the best described is the β-adrenergic stimulation, which contributes to the positive inotropic effects of catecholamines. To date, three β-adrenergic receptors (β-ARs), respectively β1-AR, β2-AR, and β3-AR, have been cloned,2 and this major achievement has led to the 2012 Nobel prize award to Robert Lefkowitz and Brian Kobilka for paving the road to the current understanding of their structures and functions. The classical pathway for β-AR receptor signaling is activation of adenylyl cyclases via Gαs, resulting in increased intracellular cyclic adenosine monophosphate (cAMP) levels. The primary target of cAMP is the cAMP-dependent protein kinase (PKA) that in turn phosphorylates the CaV1.2 channels among other key proteins of the excitation-contraction coupling (Figure 37-1). Although direct modulation by G proteins of LTCCs was first suggested to partially mediate the upregulation of ICa,L upon β-AR activation, it has been clearly established that a cAMP phosphorylation mediated by PKA is responsible for this increase.3,4 Figure 37-1 β-Adrenergic modulation of cardiac L-type calcium channels. Modulation of cardiac CaV1.2 channels by β-adrenergic receptor (β-AR) stimulation occurs via a G protein pathway. β-ARs couple to heterodimeric G proteins. Agonist catecholamine binding to β-AR activates the stimulatory heterotrimeric G protein by inducing the exchange of guanosine diphosphate (GDP) for guanosine-5′-triphosphate (GTP) on GαS. GαS dissociates from its Gβγ partner to stimulate the adenylyl cyclase (AC) present in the plasma membrane. The AC catalyses the conversion of adenosine triphosphate (ATP) in cyclic 3′,5′ monophosphate (cAMP) that in turn activates the cAMP dependent protein kinase (PKA) to phosphorylate CaV1.2. This chapter reviews the literature on the β-AR regulation of LTCCs, with emphasis on recent information on the molecular mechanisms of PKA regulation of CaV1.2 channels and the local compartmentation of β-AR/cAMP/PKA signaling around these channels. We conclude with an overview of such modulation in a pathologic context. Additional details concerning LTCCs can be found in Chapter 2. LTCCs display three distinct gating modes upon depolarization: mode 1 corresponds to brief openings, mode 2 corresponds to long lasting openings, and mode 0 corresponds to a silent mode because of unavailability.5 PKA phosphorylation of CaV1.2 channels results in a shift of the channel from the gating mode 1 to mode 2.6 As a result, ICa,L density is increased twofold to threefold, and its voltage dependence shifted slightly toward hyperpolarized potentials (Figure 37-2, A). Voltage steady-state activation and inactivation in adult mouse ventricular myocytes are presented in Figure 37-2. Activation for ICa,L starts at –40 mV and is maximal around 5 mV while inactivation begins at –45 mV and is maximal at approximately 0 mV. The overlap of the two curves defines a “window current” between –40 and 0 mV (i.e., near the action potential plateau phase). The β-adrenergic stimulation leads to an increased window current because of the effect of PKA phosphorylation on channel activation. As presented in Figure 37-2, B, isoproterenol application at a maximal concentration of 100 nM shifts the activation by 5 mV toward negative potentials, whereas a minor shift of availability of approximately 2.5 mV is observed. During maintained depolarizations, ICa,L decreases with time, a phenomenon named inactivation, which depends on time, voltage, and intracellular calcium.7 Because β-AR activation enhances calcium entry, it also accelerates ICa,L inactivation,8 and calcium-dependent inactivation becomes the main mechanism by which the channel inactivates.9 Overall, β-AR stimulation promotes ICa,L and thus the calcium entry during action potential that is partially responsible for its positive inotropic effects. Figure 37-2 The effect of β-adrenergic stimulation on ICa,L in adult mice ventricular cardiomyocytes. A, Example of current traces of ICa,L recorded using the patch-clamp technique under whole-cell conditions at 0 mV (holding potential at –50 mV) obtained before and after activation of the β-adrenergic receptors with 100 nM isoproterenol. An approximately twofold increase of ICa,L amplitude is induced by the application of the agonist. B, Influence of β-adrenergic stimulation on cardiac calcium channel activation and availability: ICa,L availability and steady-state inactivation were measured by applying a 200-ms pulse from –60 to 60 mV, followed by a 3-ms repolarization to –50 mV, before a 200-ms test pulse to 0 mV. The present curves are mean data obtained from 16 different adult mice ventricular myocytes before and after application of 100 nM isoproterenol. Steady-state inactivation curves were obtained by normalizing the peak current at each test potential to the maximal current. Activation curves were derived from current–voltage relations and described by: G / Gmax = 1 / (1+exp[{V½,act − V} / k]). A and B were obtained in similar recording conditions as described by Leroy et al.91 The LTCC current ICa,L in the working myocardium is mediated by the predominantly expressed CaV1.2 channels. These channels are multimeric proteins composed of a central subunit, α1C, the pore-forming subunit, which determines the main biophysical and pharmacologic properties of the channel. This subunit is a protein of approximately 240 kDa with 24 transmembrane segments according to its hydropathic profile, organized into four repeated domains (I to IV) of six transmembrane segments each (1 to 6), with intracellular N- and a large C-termini.4 Like other high-voltage–gated calcium channels, CaV1.2 associates with a largely extracellular disulfide-linked α2-δ subunit of 170 kDa.10 It also binds CaVβ subunits to its α interaction domain present in its intracellular I-II loop via their guanylate kinase–like domain.11 Four genes encode four CaVβ subunits (β1-4), but CaVβ2 is thought to be the main isoform expressed in the heart.12 Both α2-δ13, and CaVβ14 auxiliary subunits influence the biophysical properties and increase the trafficking of the channel at the plasma membrane. γ(4,6-8) subunits are also expressed in ventricular cells and, like γ1 for the skeletal muscle calcium channel CaV1.1, can interact with the CaV1.2 channel to modulate its function when coexpressed in HEK-293 cells; however, the exact role for these accessory proteins on CaV1.2 in native cardiac tissues remains to be determined.15 If the main effects of β-AR stimulation on voltage dependence and amplitude of the ICa,L and their consequences for the fight-or-flight response are well documented in the literature, the molecular events that mediate the increase of CaV1.2 activity during a sympathetic stimulation remain elusive; this is due to the difficulty of reconstituting such modulation in heterologous overexpression systems. The α1C-subunit of CaV1.2 channels exhibits multiple potential PKA phosphorylation sites in the N- and the C-terminal regions (Figure 37-3).2 Despite the fact that α1C is a substrate for phosphorylation by PKA in vitro,16,17 several attempts to mimic the adrenergic stimulation of CaV1.2 channels in expression systems have failed.18–20 This failure led to the hypothesis that at least one missing link in heterologous systems would preclude the reconstitution of such modulation of overexpressed CaV1.2 channels. One of these links could be an A-kinase-anchoring protein (AKAP) that allows the cAMP modulation and the PKA phosphorylation of serine 1928 in the C-terminus of CaV1.2 channels in HEK293 cells.21 A conserved leucine zipper motif in the C-terminus of CaV1.2 identified in native cardiac cells directly anchors a low molecular weight AKAP15-PKA complex to ensure a fast and efficient β-adrenergic modulation of ICa,L (see Figure 37-3),22 allowing its phosphorylation at serine 1928 when β-ARs are stimulated.23 Surprisingly, the C-terminal part of rabbit CaV1.2 undergoes proteolytic processing by calpain at residue 1821, leaving two size forms of the α1C-subunit of these channels expressed in cardiac cells,17 whereas this distal 37 to 50-kDa peptide contains the serine 1928 phosphorylated by PKA and the binding site for AKAP15. In fact, this fragment constitutes a potent autoinhibitory domain that covalently associates with the proximal C-terminal part of α1C, reducing its open probability and shifting the voltage dependence of activation to depolarized potentials.24 The role of serine 1928 in ICa,L upregulation by β-AR has since been challenged. A first study showed that a DHP-resistant CaV1.2 channels mutated at position 1928 (S1928A) displays an unaltered response to the β-AR agonist isoproterenol when overexpressed in isolated ventricular myocytes.25 Moreover, the generation of a S1928A knock-in mouse model confirmed that the phosphorylation of this residue by PKA is not required for the functional effects of β-AR stimulation on CaV1.2 in cardiac cells, because these mice exhibit an essentially conserved response of ICa,L to isoproterenol and typical chronotropic and inotropic responses to β-AR stimulation.26 Therefore, although phosphorylation of serine 1928 within the C-terminus of CaV1.2 definitely occurs when β-ARs are stimulated,17,21,23 it does not correlate with the functional effects observed on ICa,L. As a result, PKA would phosphorylate other PKA sites within the channel or another binding partner to mediate the increase of its activity. Figure 37-3 Subunit structure of cardiac L-type CaV1.2 channel and phosphorylation sites for PKA mediated upregulation of ICa,L. The α1C-subunit constitutes the pore-forming subunit of the cardiac CaV1.2 channel. It presents four domains (I to IV) composed of six transmembrane segments (S1 to S6), with S4 exhibiting positively charged residues that confer their sensitivity to potential and a pore loop between S5 and S6 of each domains. The intracellular I-II loop of the α1C-subunit associates with the β interaction domain (BID) in the guanylate kinase–like domain of the intracellular CaVβ2a auxiliary subunit via its α interaction domain (AID). CaV1.2 associates also with the largely extracellular glycosylphosphatidylinositol (GPI)-anchored α2-δ subunit. The C-terminal part of the α1C undergoes proteolytic cleavage by calpain at residue 1821. The cleaved distal fragment associates monocovalently with the proximal C-terminal part to inhibit the channel. This necessary association for β-AR regulation of CaV1.2 is relieved by the phosphorylation of serine 1700 by the PKA tethered in the distal C-terminus by AKAP15 to produce the upregulation of channel activity. PKA also phosphorylates serine 1704 to control its basal activity while phosphorylation of serine 1928 serves an undetermined regulatory function. PKA also phosphorylates CaVβ2a on the two serine residues 478 and 479. This subunit associates to the giant ahnak cytosqueletal protein that could also be an important factor for the cAMP/PKA regulation of CaV1.2 channels. Three serines (S459, S478, and S479) of the cardiac the CaVβ2a subunit were identified as putative PKA phosphorylation sites.27 Coexpression of this subunit with a CaV1.2 lacking the C-terminal part including the serine 1928 in TsA-201 cells allowed an upregulation of the current by cAMP/PKA pathway, whereas expression of mutated CaVβ2a at serine 478 and 479, but not at position 459, was unable to do so.28 The authors concluded that the CaVβ2a ancillary subunit was the main target for PKA to mediate the β–AR stimulation of ICa,L; however, this conclusion has been questioned by experiments realized in a more physiologic context. Overexpression of a CaVβ2a construct mutated for its PKA phosphorylation sites in ventricular cardiac cells did not prevent the cAMP/PKA modulation of ICa,L.14 Nonetheless, the CaVβ2 subunit can associate with the giant cytoskeletal protein of 700 kDa named ahnak, which emerged as an important player in the β-AR regulation of cardiac ICa,L (see Figure 37-3). Through its interaction with CaVβ2, ahnak would serve as a brake on ICa,L that would be relieved by PKA phosphorylation of the ancillary subunit and the cytoskeletal protein.29 Interestingly, ahnak polymorphism occurs, and the genetic variant generated interferes with the β-AR stimulation of ICa,L by reducing the CaVβ2 interaction with ahnak.29 However, if the involvement of the CaVβ or other binding partners such as ahnak cannot be ruled out completely, recent studies have reaffirmed the importance of the proteolytically cleaved distal C-terminus of CaV1.2 for its upregulation upon β-AR stimulation. In addition, a new model for the molecular basis of β-AR stimulation of CaV1.2 has been proposed.30 Overexpression in TsA-201 cells of a truncated CaV1.2 channel at position A1800 (the site of in vivo proteolytic cleavage previously determined for skeletal muscle CaV1.1 channel31) with α2δ1 and CaVβ1b and the distal C-terminal part of the channel (peptide from 1821-2171) produced a functional channel that is autoinhibited. Two presumed sites for PKA phosphorylation were identified upstream from the proteolytic site—a serine at position 1700 and a threonine 1704 at the interface of the distal and the proximal C-terminal parts. Although a mutation of S1928 confirmed that its phosphorylation is not required for the increase of the current, substitution of T1704 and S1700 for alanines revealed that phosphorylation of T1704 is required for basal CaV1.2 channel activity, whereas S1700 is crucial for its cAMP/PKA-induced upregulation. In this scheme, the AKAP15 is still required to coordinate the PKA phosphorylation of S1700 that disrupts the interaction of the noncovalently distal C-terminus, thus relieving the inhibition of the channel.30 This assumption is reinforced by the fact that mice expressing a CaV1.2 channel deleted for the distal C-terminus display altered cAMP/PKA regulation accompanied with reduced expression of AKAP15.32 In light of the knowledge accumulated over the years, it is evident that intracellular cAMP is not uniformly distributed, and nor is PKA uniformly activated within cardiomyocytes upon β-AR stimulation.33 On the contrary, a tight cAMP/PKA compartmentation is required for adequate processing and targeting of the information generated at the cell surface, conferring the specificity of the response to various hormones linked to Gαs-coupled receptors.34,35 In the case of β-ARs, several processes contribute to a localized cAMP/PKA response: catecholamines activate different β-AR subtypes located at different places of the cell surface (e.g., caveolae, T-tubules); Gαs activation of different adenylyl cyclase (AC) isoforms can lead to cAMP synthesis at different locations; cAMP diffusion may be restricted because of localized phosphodiesterase (PDE) activity; anchoring of PKA to AKAPs position PKA at different subcellular compartments to selectively phosphorylate a local pool of proteins for specific cellular processes.36,37 In addition, AKAPs also ensure that PKA is coupled to its upstream activators, including membrane β-ARs and ACs, and to signal termination enzymes such as PDEs and phosphatases.36 Although only three types of β-adrenergic receptors (β-ARs) have been cloned (β1-, β2-, and β3-ARs), the effect of catecholamines in the human heart is generally attributed to β1– and β2-ARs. β1– and β2-ARs are highly homologous receptors and are both positively coupled to AC/cAMP/PKA cascade, ICa,L and cardiac performance33; however, they exert opposite effects on hypertrophy38,39 and apoptosis,40,41 and their respective contribution varies significantly depending on the cardiac tissue, pathophysiologic state, age, or developmental stage.42 Part of the difference is because β2-ARs couple not only to Gαs but also to Gαi proteins, and this confines the cAMP-dependent signal to the membrane compartment and to activation of the LTCCs.43 Another difference between the two β-AR subtypes is their location at the cell surface, with β2-ARs present in the caveolae/lipid rafts44,45 of the transverse-tubular structure46 and β1-ARs distributed throughout both caveolae–lipid rafts and nonlipid raft membrane domains44 and in both plasma and T-tubular membranes.46 Accordingly, the β2-AR downstream activation of ICa,L is sensitive to disruption of caveolae by cholesterol depletion, whereas the β1-AR stimulatory effect is not.47 Therefore, β2-AR stimulation exerts a local activation of LTCCs, whereas β1-AR stimulation leads to activation of LTCCs in the distance.43,48,49 The β3-AR differs from β1-and β2-AR subtypes in its molecular structure and pharmacologic functions.50 Expression of β3-AR was demonstrated in human myocardium at the mRNA51,52 and protein levels.52–55 Interestingly, β3-AR activation produces a negative inotropic effect in human endomyocardial biopsies from transplanted hearts50,51 and in left ventricular samples from failing and nonfailing explanted hearts50,54 that is due to activation of the NO/cGMP pathway, but increases ICa,L and contractility in human atrial tissue via the cAMP/PKA pathway (Figure 37-4).56 This effect is reminiscent of the contractile effects of the serotonin 5-HT4 receptors,57 which are also coupled to increases in force of contraction58 or ICa,L59 in atria but not in healthy ventricles. Figure 37-4 Opposite effect of β3-adrenergic receptor stimulation on L-type calcium channel (LTCC) activity in a human atrium and ventricle. In the atrium (top), the β3-adrenergic receptor (β3-AR) is positively coupled to the Gs-protein and to adenylyl cyclase (AC). Upon activation of the β3-AR, cAMP synthesis is increased, which leads to PKA phosphorylation of LTCCs and a stimulation of Ca2+ influx. In the human ventricle (bottom), the β3-AR is coupled to the endothelial nitric oxide synthase (eNOS), presumably via the Gi/o-protein. Activation of eNOS leads to NO production, which activates soluble guanylyl cyclase (sGC) and increases intracellular cGMP concentration. This leads to PKG-phosphorylation of LTCCs and an inhibition of Ca2+ influx. Cyclic AMP signaling components are organized into multiprotein complexes, an arrangement that increases both efficiency and specificity of the transduction cascade. AKAPs have an essential role in these arrangements. AKAPs form a large family of proteins comprising more than 50 members whose primary function is to anchor PKA in the vicinity of its substrates, thus ensuring the preferential phosphorylation of a limited number of targets.60,61 As discussed earlier, AKAP15 (also called AKAP 7 or AKAP15/18) is the main AKAP controlling the PKA phosphorylation of LTCCs. However, in a recent study, CaV1.2 phosphorylation and β-AR stimulation of ICa,L was found to be unchanged in mice in which the gene encoding this protein was inactivated, suggesting that PKA is anchored by a different protein to the channel.62 Furthermore, AKAP5 was found to target ACs, PKA and phosphatases within caveolae to allow specific PKA phosphorylation of the subpopulation of channels present in this compartment upon β-AR stimulation.63 Although AKAPs share in common their ability to bind PKA, they are remarkably diverse scaffold proteins. Within each signalosome, AKAPs couple PKA to different substrates, enhancing the rate and fidelity of their phosphorylation by the kinase. Importantly, AKAPs not only bind PKA but act as scaffold proteins for other signaling components, including phosphatases 1 and 2,64,65 Epac,66 adenylyl cyclases,67,68 and PDEs.69 Recently, it has been demonstrated that the phosphoinositide 3-kinase p110γ, that was shown recently tether PKA,70 orchestrates multiprotein complexes including different PDEs to control cardiac CaV1.2 phosphorylation during β-AR stimulation.71 The combination of PDEs and phosphatases present in individual AKAP complexes will affect the duration, amplitude, and spatial extent of cAMP/PKA signaling. Thus, by bringing together different combinations of upstream and downstream signaling molecules, AKAPs provide the architectural infrastructure for specialization of the cAMP signaling network.61,66,72 Localized cAMP signals can be generated by the interplay between discrete cAMP production sites and restricted diffusion within the cytoplasm. Restricted diffusion of cAMP can be achieved by several means. A first possibility is that physical barriers are created by specialized membrane structures within the cytoplasm. This method was initially proposed to explain the differences in cAMP concentration elicited by PGE1 at the plasma membrane and in the bulk cytosol of HEK293 cells, although an experimental proof that this actually occurs is still lacking.73 Another important mechanism that limits cAMP diffusion is cAMP degradation by PDEs, which appears to be critical to the formation of dynamic microdomains that confer specificity of the response.35,60 Cardiac cAMP PDEs degrading belong to five families (PDE1 to PDE4 and PDE8) that can be distinguished by distinct enzymatic properties and pharmacology.74 Among these families various enzymes were shown to degrade cAMP to allow a fine tuning of ICa,L regulation by PKA in the heart. Although PDE2 is not highly expressed in cardiomyocytes, it controls LTCC activity in various species, including human atrial myocytes.75–78 This enzyme is activated by cGMP, and stimulation of guanylyl cyclase strongly decreases local cAMP levels controlling ICa,L with only modest effects on its global concentration, suggesting the existence of a cAMP microdomain including β-AR and LTCCs under tight control of PDE2.79 Contrary to PDE2, PDE3 is inhibited by cGMP; this explains in part why cGMP at low concentration can also increase basal ICa,L as shown in human atrial myocytes.78,80 In rodents, PDE3 and PDE4 are the major contributors to the total cAMP-hydrolytic activity34,81 and PDE4 is dominant to modulate β-AR regulation of cAMP levels.49,82–84 Multiple PDE4 variants associate with β-ARs,85–87 RyR2,88 SERCA2,89,90 ICa,L,91 and IKs72 to exert local control of ECC. In larger mammals, PDE3 activity is dominant in microsomal fractions,92–94 and PDE3 inhibitors exert a potent positive inotropic effect.95 Selective inhibition of PDE3 with milrinone has been shown to improve cardiac contractility in patients with congestive heart failure.96 The role of PDE4 is less well defined, but evidence is emerging that PDE4 could also have an important role in these species. In the canine heart, a large PDE4 activity is found in the cytoplasm,92 but PDE4 is also present in microsomal fractions, where it accounts for approximately 20% of the activity.93 Recent studies have indicated that PDE4 is expressed in the human ventricle where, similar to rodents, it associates with β-ARs, RyR2, and phospholamban.81,88 Moreover, PDE4 is the main PDE modulating LTCC activity in rodent cardiomyocytes,77,84 and PDE4 was recently shown to control ICa,L, ECC and arrhythmias in the human atrium.97 Early evidence of the contribution of PDEs to intracellular cyclic nucleotide compartmentation was obtained by comparing the effects of the nonselective β-AR agonist Iso, or the nonselective PDE inhibitor, 3-isobutyl-1-methylxanthine (IBMX), or the PDE3 inhibitor milrinone in guinea pig perfused hearts. Whereas each of these treatments increased intracellular cAMP and produced positive inotropic and lusitropic effects, differences in the phosphorylation pattern of PLB, TnI, and MyBP-C by PKA were observed.98 These results were attributed to a functional cellular compartmentation of cAMP and PKA substrates owing to a different expression of PDEs at the membrane and in the cytosol.92 In canine ventricular myocytes, an increase in particulate but not total cAMP correlated to an increase in Ca2+ transient amplitude and decay kinetics.99 In response to β-AR stimulation, approximately 45% of the total cAMP was found in the particulate fraction, but this fraction declined to less than 20% when IBMX was added to Iso, although cAMP production was up to threefold to fourfold greater. These results show that cAMP-PDEs reside predominantly in the cytoplasm, where they prevent excessive cAMP accumulation upon β-AR activation. Thus, PDEs appear to be important to maintain the specificity of the β-AR response by limiting the amount of cAMP diffusing from membrane to cytoplasm. Similar results were obtained when studying the effect of PDE inhibition on ICa,L
CaV1.2 and β-Adrenergic Regulation of Cardiac Function
Overview of the β-Adrenergic Regulation of L-Type Calcium Channels
Molecular Mechanisms of Protein Kinase Regulation of L-Type Calcium Channels
α-1 Subunit
β-Subunit
Compartmentation of cAMP/PKA Regulation of L-Type Calcium Channels
Receptor Specificity
Role of A-Kinase-Anchoring Proteins
Role of Phosphodiesterases
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


CaV1.2 and β-Adrenergic Regulation of Cardiac Function
