OBJECTIVES
The student understands the primary disturbances, compensatory responses, decompensatory processes, and possible therapeutic interventions that pertain to various abnormal cardiovascular situations:
![]() Defines circulatory shock.
Defines circulatory shock.
![]() Identifies the primary disturbances that can account for cardiogenic, hypovolemic, anaphylactic, septic, and neurogenic shock states.
Identifies the primary disturbances that can account for cardiogenic, hypovolemic, anaphylactic, septic, and neurogenic shock states.
![]() Lists the compensatory processes that may arise during various types of circulatory shock.
Lists the compensatory processes that may arise during various types of circulatory shock.
![]() Identifies the decompensatory processes that may arise during shock and describes how these lead to irreversible shock states.
Identifies the decompensatory processes that may arise during shock and describes how these lead to irreversible shock states.
![]() Indicates how coronary artery disease may lead to abnormal cardiac function.
Indicates how coronary artery disease may lead to abnormal cardiac function.
![]() Defines the term angina pectoris and describes the mechanisms that promote its development.
Defines the term angina pectoris and describes the mechanisms that promote its development.
![]() Indicates the mechanisms by which various therapeutic interventions may alleviate angina and myocardial ischemia in association with coronary artery disease.
Indicates the mechanisms by which various therapeutic interventions may alleviate angina and myocardial ischemia in association with coronary artery disease.
![]() Defines the term heart failure and differentiates between acute and chronic heart failure and between systolic and diastolic failure.
Defines the term heart failure and differentiates between acute and chronic heart failure and between systolic and diastolic failure.
![]() Identifies the short-term and long-term compensatory processes that accompany chronic systolic heart failure.
Identifies the short-term and long-term compensatory processes that accompany chronic systolic heart failure.
![]() Describes the advantages and disadvantages of the fluid accumulation that accompanies systolic heart failure.
Describes the advantages and disadvantages of the fluid accumulation that accompanies systolic heart failure.
![]() Defines pulmonary and systemic arterial hypertension.
Defines pulmonary and systemic arterial hypertension.
![]() Identifies the various factors that may contribute to the development of systemic hypertension.
Identifies the various factors that may contribute to the development of systemic hypertension.
![]() Describes the role of the kidney in establishing and/or maintaining systemic hypertension.
Describes the role of the kidney in establishing and/or maintaining systemic hypertension.
In this last chapter, some of the pathological situations that can interfere with the homeostatic functions of the cardiovascular system are introduced. It is not intended as an in-depth coverage of cardiovascular diseases but rather as an introductory presentation of how the physiological processes described previously are evoked and/or altered during various abnormal cardiovascular states. In each case, there is generally a primary disturbance that evokes appropriate compensatory reflex responses. Often, however, pathological situations also lead to inappropriate “decompensatory processes,” which tend to accelerate the deterioration of cardiovascular function. Therapeutic interventions may be required and are often designed to limit or reverse these decompensatory processes. Students are again encouraged to review the summary of cardiovascular variables and their determinants in Appendix C because a thorough knowledge of this material will greatly help to understand the physiological consequences of these abnormalities.
CIRCULATORY SHOCK
![]() A state of circulatory “shock” exists whenever there is a generalized, severe reduction in blood supply to the body tissues and the metabolic needs of the tissues are not met. Even with all cardiovascular compensatory mechanisms activated, arterial pressure is usually (though not always) low in shock. In severe shock states of any etiology, inadequate brain blood flow leads to loss of consciousness often with sudden onset (called syncope). The approach to understanding the causes and selecting an appropriate treatment depends on determination of the underlying primary disturbance. Recall that arterial pressure is determined by cardiac output and total peripheral resistance, so any loss in blood pressure is a result of a decrease in either one or both of these variables.
A state of circulatory “shock” exists whenever there is a generalized, severe reduction in blood supply to the body tissues and the metabolic needs of the tissues are not met. Even with all cardiovascular compensatory mechanisms activated, arterial pressure is usually (though not always) low in shock. In severe shock states of any etiology, inadequate brain blood flow leads to loss of consciousness often with sudden onset (called syncope). The approach to understanding the causes and selecting an appropriate treatment depends on determination of the underlying primary disturbance. Recall that arterial pressure is determined by cardiac output and total peripheral resistance, so any loss in blood pressure is a result of a decrease in either one or both of these variables.
Primary Disturbances
![]() In general, the shock state is precipitated by one of three cardiovascular crises: (1) severely depressed myocardial functional ability, (2) grossly inadequate cardiac filling due to low mean circulatory filling pressure, or (3) profound systemic vasodilation either due to the abnormal presence of powerful vasodilators or due to the absence of neurogenic tone normally supplied by the sympathetic nervous system. The consequences of these primary disturbances are represented in the five categories of shock summarized in Figure 11–1.
In general, the shock state is precipitated by one of three cardiovascular crises: (1) severely depressed myocardial functional ability, (2) grossly inadequate cardiac filling due to low mean circulatory filling pressure, or (3) profound systemic vasodilation either due to the abnormal presence of powerful vasodilators or due to the absence of neurogenic tone normally supplied by the sympathetic nervous system. The consequences of these primary disturbances are represented in the five categories of shock summarized in Figure 11–1.
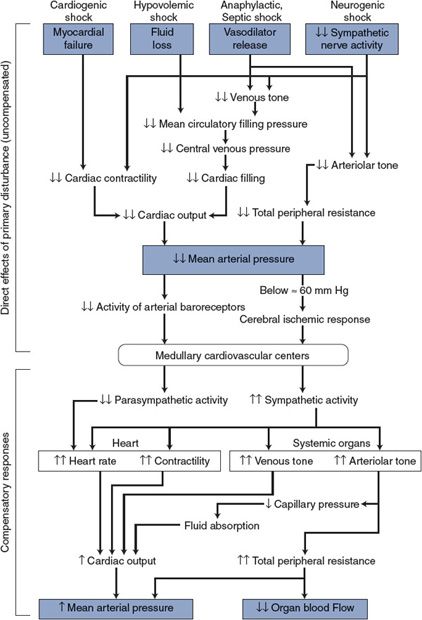
Figure 11–1. Cardiovascular alterations in shock.
1. Cardiogenic shock occurs whenever cardiac pumping ability is compromised (eg, as a result of severe arrhythmias, abrupt valve malfunction, coronary occlusions, or myocardial infarction). The direct consequence of any of these abnormalities is a significant fall in cardiac output.
2. Hypovolemic shock accompanies significant hemorrhage (usually greater than 20% of blood volume), or fluid loss from severe burns, chronic diarrhea, or prolonged vomiting. These situations can induce shock by depleting body fluids and thus circulating blood volume. The direct consequence of hypovolemia is inadequate cardiac filling and reduced stroke volume.
There are some situations that may result in reduced cardiac filling that are not related to hypovolemia. For example, cardiac tamponade, associated with fluid accumulation in the pericardial sac, prevents adequate diastolic filling as does the occurrence of a pulmonary embolus (a clot mobilized from systemic veins lodging in a pulmonary vessel).
3. Anaphylactic shock occurs as a result of a severe allergic reaction to an antigen to which the patient has developed a sensitivity (eg, insect bites, antibiotics, and certain foods). This immunological event, also called an “immediate hypersensitivity reaction,” is mediated by several substances (such as histamine, prostaglandins, leukotrienes, and bradykinin) that, by multiple mechanisms, results in substantial arteriolar vasodilation, increases in microvascular permeability, and loss of peripheral venous tone. These combine to reduce both total peripheral resistance and cardiac output.
4. Septic shock is also caused by profound vasodilation but specifically from substances released into the circulating blood by infective agents. One of the most common is endotoxin, a lipopolysaccharide released from bacteria. This substance induces the formation of a nitric oxide synthase (called inducible nitric oxide synthase to distinguish it from the normally present constitutive nitric oxide synthase) in endothelial cells, vascular smooth muscle, and macrophages that then produce large amounts of the potent vasodilator nitric oxide. The term distributive shock is sometimes used to describe both the anaphylactic and septic shock states.
5. Neurogenic shock is produced by loss of vascular tone due to inhibition of the normal tonic activity of the sympathetic vasoconstrictor nerves and often occurs with deep general anesthesia or in reflex response to deep pain associated with traumatic injuries. It may also be accompanied by an increase in vagal activity, which significantly slows the cardiac beating rate. This type of shock is often referred to a vasovagal syncope. The transient syncope evoked by strong emotions is a mild form of neurogenic shock and is usually quickly reversible.
As shown in the top half of Figure 11–1, the common primary disturbances in all forms of shock are decreased cardiac output and/or total peripheral resistance leading to decreased mean arterial pressure. Generally, the reduction in arterial pressure is substantial, and so therefore, is the influence on the cardiovascular centers from reduced arterial baroreceptor discharge rate. In addition, in the case of hypovolemic, anaphylactic, and septic shock, diminished activity of the cardiopulmonary baroreceptors due to a decrease in central venous pressure and/or volume acts on the medullary cardiovascular centers to stimulate sympathetic output.1 If arterial pressure falls below approximately 60 mm Hg, brain blood flow begins to fall and this elicits the cerebral ischemic response. As indicated in Chapter 9, the cerebral ischemic response causes intense activation of the sympathetic nerves.
Compensatory Mechanisms
![]() In general, the various forms of shock evoke the compensatory responses in the autonomic nervous system that we would expect from a fall in blood pressure.2 These increases in sympathetic activity and decreases in parasympathetic activity are indicated in the bottom half of Figure 11–1. Cardiac and peripheral vascular compensatory responses to shock, however, may be much more intense than those that accompany more ordinary cardiovascular disturbances. Many of the commonly recognized symptoms of shock (eg, pallor, cold clammy skin, rapid heart rate, muscle weakness, and venous constriction) are a result of greatly increased sympathetic nerve activity. When the immediate compensatory processes are inadequate, the individual may also show signs of abnormally low arterial pressure and reduced cerebral perfusion, such as dizziness, confusion, or loss of consciousness.
In general, the various forms of shock evoke the compensatory responses in the autonomic nervous system that we would expect from a fall in blood pressure.2 These increases in sympathetic activity and decreases in parasympathetic activity are indicated in the bottom half of Figure 11–1. Cardiac and peripheral vascular compensatory responses to shock, however, may be much more intense than those that accompany more ordinary cardiovascular disturbances. Many of the commonly recognized symptoms of shock (eg, pallor, cold clammy skin, rapid heart rate, muscle weakness, and venous constriction) are a result of greatly increased sympathetic nerve activity. When the immediate compensatory processes are inadequate, the individual may also show signs of abnormally low arterial pressure and reduced cerebral perfusion, such as dizziness, confusion, or loss of consciousness.
Additional compensatory processes initiated during the shock state may include the following:
1. Rapid and shallow breathing occurs, which promotes venous return to the heart by action of the respiratory pump.
2. Increased renin release from the kidney as a result of sympathetic stimulation promotes the formation of the hormone, angiotensin II, which is a potent vasoconstrictor and participates in the increase in total peripheral resistance even in mild shock states.
3. Increased circulating levels of vasopressin (also known as antidiuretic hormone) from the posterior pituitary gland contribute to the increase in total peripheral resistance. This hormone is released in response to decreased firing of the cardiopulmonary and arterial baroreceptors.
4. Increased circulating levels of epinephrine from the adrenal medulla in response to sympathetic stimulation contribute to systemic vasoconstriction.
5. Reduced capillary hydrostatic pressure resulting from intense arteriolar constriction reduces capillary hydrostatic pressure and promotes fluid movement from the interstitial space into the vascular space.
6. Increased glycogenolysis in the liver induced by epinephrine and norepinephrine results in a release of glucose and a rise in blood (and interstitial) glucose levels and, more importantly, a rise in extracellular osmolarity by as many as 20 mOsm. This will induce a shift of fluid from the intracellular space into the extracellular (including intravascular) space.
The latter two processes result in a sort of “autotransfusion” that can move as much as a liter of fluid into the vascular space in the first hour after the onset of the shock episode. This fluid shift accounts for the reduction in hematocrit that is commonly observed in hemorrhagic shock. The extent of fluid shift may be limited by a reduction in colloid osmotic pressure.
In addition to the immediate compensatory responses shown in Figure 11–1, fluid retention mechanisms are evoked by hypovolemic states that affect the situation in the long term. The production and release of the antidiuretic hormone (vasopressin) from the posterior pituitary promote water retention by the kidneys. Furthermore, activation of the renin–angiotensin–aldosterone pathway promotes renal sodium retention (via aldosterone) and the thirst sensation and drinking behavior (via angiotensin II). These processes contribute to the replenishment of extracellular fluid volume within a few days of the shock episode.
Decompensatory Processes
![]() Often the strong compensatory responses during shock evoked by the intense sympathetic activation are capable of preventing drastic reductions in arterial pressure. However, because the compensatory mechanisms involve overwhelming arteriolar vasoconstriction, perfusion of tissues other than the heart and the brain may be inadequate despite nearly normal arterial pressure. For example, blood flow through vital organs such as the liver, gastrointestinal tract, and kidneys may be reduced nearly to zero by intense sympathetic activation. The possibility of permanent renal, hepatic, or GI tract ischemic damage is a very real concern even in seemingly mild shock situations. Patients who have apparently recovered from a state of shock may die several days later because of renal failure, uremia, or sepsis due to bacterial penetration of the weakened mucosal barrier in the GI tract.
Often the strong compensatory responses during shock evoked by the intense sympathetic activation are capable of preventing drastic reductions in arterial pressure. However, because the compensatory mechanisms involve overwhelming arteriolar vasoconstriction, perfusion of tissues other than the heart and the brain may be inadequate despite nearly normal arterial pressure. For example, blood flow through vital organs such as the liver, gastrointestinal tract, and kidneys may be reduced nearly to zero by intense sympathetic activation. The possibility of permanent renal, hepatic, or GI tract ischemic damage is a very real concern even in seemingly mild shock situations. Patients who have apparently recovered from a state of shock may die several days later because of renal failure, uremia, or sepsis due to bacterial penetration of the weakened mucosal barrier in the GI tract.
The immediate danger with shock is that it may enter the progressive stage, wherein the general cardiovascular situation progressively degenerates, or, worse yet, enter the irreversible stage, where no intervention can halt the ultimate collapse of cardiovascular system that results in death.
The mechanisms behind progressive and irreversible shock are not completely understood. However, it is clear from the mechanisms shown in Figure 11–2 that bodily homeostasis can progressively deteriorate with prolonged reductions in organ blood flow. These homeostatic disturbances, in turn, adversely affect various components of the cardiovascular system so that arterial pressure and organ blood flow are further reduced. Note that the events shown in Figure 11–2 are decompensatory mechanisms. Reduced arterial pressure leads to alterations that further reduce arterial pressure rather than correct it (ie, a positive feedback process). These decompensatory mechanisms that are occurring at the tissue level to lower blood pressure are eventually further compounded by a reduction in sympathetic drive and a change from vasoconstriction to vasodilation with a further lowering of blood pressure. The factors that lead to this unexpected reduction in sympathetic drive from the medullary cardiovascular centers are not clearly understood. If the shock state is severe enough and/or has persisted long enough to enter the progressive stage, the self-reinforcing decompensatory mechanisms progressively drive arterial pressure down. Unless corrective measures are taken quickly, death will ultimately result.
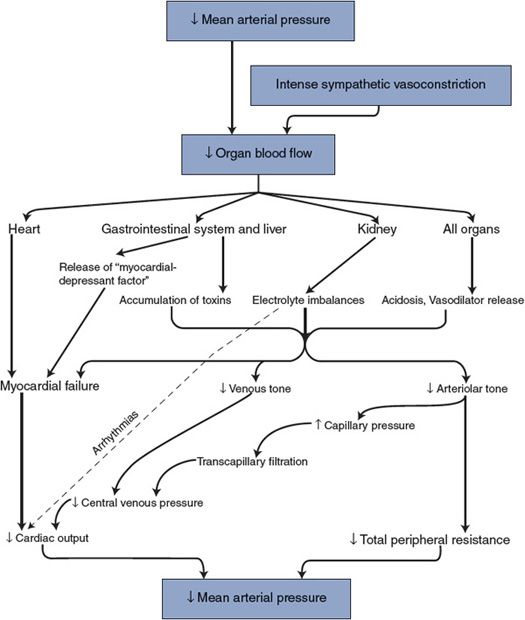
Figure 11–2. Decompensatory mechanisms in shock.
CARDIAC DISTURBANCES
Coronary Artery Disease
DEFINITION AND PHYSIOLOGICAL CONSEQUENCES
![]() Whenever coronary blood flow falls below that required to meet the metabolic needs of the heart, the myocardium is said to be ischemic and the pumping capability of the heart is impaired. The most common cause of myocardial ischemia is atherosclerotic disease of the large coronary arteries. In atherosclerotic disease, localized lipid deposits called plaques develop within the arterial walls. With severe disease, these plaques may become calcified and so large that they physically narrow the lumen of arteries (producing a stenosis) and thus greatly and permanently increase the normally low vascular resistance of these large arteries. This extra resistance adds to the resistance of other coronary vascular segments and tends to reduce coronary flow. If the coronary artery stenosis is not too severe, local metabolic vasodilator mechanisms may reduce arteriolar resistance sufficiently to compensate for the abnormally large arterial resistance. Thus, an individual with coronary artery disease may have perfectly normal coronary blood flow when resting. A coronary artery stenosis of any significance will, however, limit the extent to which coronary flow can increase above its resting value by reducing maximum achievable coronary flow. This occurs because, even with very low arteriolar resistance, the overall vascular resistance of the coronary vascular bed is high if resistance in the large arteries is high.
Whenever coronary blood flow falls below that required to meet the metabolic needs of the heart, the myocardium is said to be ischemic and the pumping capability of the heart is impaired. The most common cause of myocardial ischemia is atherosclerotic disease of the large coronary arteries. In atherosclerotic disease, localized lipid deposits called plaques develop within the arterial walls. With severe disease, these plaques may become calcified and so large that they physically narrow the lumen of arteries (producing a stenosis) and thus greatly and permanently increase the normally low vascular resistance of these large arteries. This extra resistance adds to the resistance of other coronary vascular segments and tends to reduce coronary flow. If the coronary artery stenosis is not too severe, local metabolic vasodilator mechanisms may reduce arteriolar resistance sufficiently to compensate for the abnormally large arterial resistance. Thus, an individual with coronary artery disease may have perfectly normal coronary blood flow when resting. A coronary artery stenosis of any significance will, however, limit the extent to which coronary flow can increase above its resting value by reducing maximum achievable coronary flow. This occurs because, even with very low arteriolar resistance, the overall vascular resistance of the coronary vascular bed is high if resistance in the large arteries is high.
Coronary artery disease can jeopardize cardiac function in several ways. Ischemic muscle cells are electrically irritable and unstable, and the danger of developing cardiac arrhythmias and fibrillation is enhanced. During ischemia, the normal cardiac electrical excitation pathways may be altered and often ectopic pacemaker foci develop. Electrocardiographic manifestations of myocardial ischemia can be observed in individuals with coronary artery disease during exercise stress tests. In addition, there is evidence that platelet aggregation and clotting function may be abnormal in atherosclerotic coronary arteries and the danger of thrombus or emboli formation is enhanced. It appears that certain platelet suppressants or anticoagulants such as aspirin may be beneficial in the treatment of this consequence of coronary artery disease. (Details of the blood clotting process are included in Appendix D.)
Myocardial ischemia not only impairs the pumping ability of the heart but also produces intense, debilitating chest pain called angina pectoris. Anginal pain is often absent in individuals with coronary artery disease when they are resting but is induced during physical exertion or emotional excitement. Both of these situations elicit an increase in sympathetic tone that increases myocardial oxygen consumption. Myocardial ischemia and chest pain will result if coronary blood flow cannot keep pace with the increase in myocardial metabolism.
DIAGNOSIS
Coronary artery imaging techniques (described in Chapter 4) have proven very useful at determining the extent of coronary artery disease. For example, calcification of plaques (which is a significant indicator of advanced atherosclerosis) can be assessed noninvasively with specialized CT scans or magnetic resonance imaging. Specific information about the site(s) and degree of narrowing of the major coronary vessels can also be obtained invasively by angiography with injection of a radioopaque dye directly into the coronary arteries.
TREATMENTS
Primary treatment of chronic coronary artery disease (and atherosclerosis, in general) should include attempts to lower blood lipids by dietary and pharmacological techniques that prevent (and possibly reverse) further development of the plaques. The interested student should consult medical biochemistry and pharmacology texts for a complete discussion of this very important topic.
Treatment of angina that is a result of coronary artery disease may involve several different pharmacological approaches. First, quick-acting vasodilator drugs such as nitroglycerin may be used to provide primary relief from an anginal attack. These drugs may act directly on coronary vessels to acutely increase coronary blood flow. In addition to increasing myocardial oxygen delivery, nitrates may reduce myocardial oxygen demand by dilating systemic veins, which reduces venous return, central venous filling, and cardiac preload, and by dilating systemic arterioles, which reduces arterial resistance, arterial pressure, and cardiac afterload. Second, β-adrenergic blocking agents such as propranolol may be used to block the effects of cardiac sympathetic nerves on the heart rate and contractility. These agents limit myocardial oxygen consumption and prevent it from increasing above the level that the compromised coronary blood flow can sustain. Third, calcium channel blockers such as verapamil may be used to dilate coronary and systemic blood vessels. These drugs, which block entry of calcium into the vascular smooth muscle cell, interfere with normal excitation–contraction coupling. They have been found to be useful for treating the type of angina caused by vasoconstrictive spasms of large coronary arteries (Prinzmetal’s angina).
Invasive or surgical interventions are commonly used to eliminate a chronic coronary artery stenosis. X-ray techniques combined with radioopaque dye injections can be used to visualize a balloon-tipped catheter as it is threaded into the coronary artery to the occluded region. Rapid inflation of the balloon squeezes the plaque against the vessel wall and improves the patency of the vessel (coronary angioplasty). A small tube-like expandible device called a stent is often implanted inside the vessel at the angioplasty site. This rigid implant promotes continued patency of the vessel over a longer period than angioplasty alone. If angioplasty and stent placement are inappropriate or unsuccessful, coronary bypass surgery may be performed. The stenotic coronary artery segments are bypassed by implanting parallel low-resistance pathways formed from either natural (eg, saphenous vein or mammary artery) or artificial vessels.
Acute Coronary Occlusion—Myocardial Infarction
![]() An acute interruption in coronary blood flow is most often a result of the sudden arrival of a blood clot (eg, an embolism released from a clot in a fibrillating atrium) or the formation of an intravascular clot at the site of a ruptured atherosclerotic plaque. Either of these events may abruptly occlude or significantly narrow a major coronary artery. This is indeed a crisis situation and demands immediate attention. The physiological consequences of such an immediate occlusion are discussed in the preceding text under the topic of “Cardiogenic Shock.” Treatments may include emergency coronary angioplasty (described earlier) with the placement of a stent, which may allow immediate restoration of flow to the ischemic area. Another method for treatment of acute myocardial infarction is the intravascular injection of thrombolytic substances (eg, streptokinase or tissue plasminogen-activating factors) that dissolve blood clots. This approach is most successful when these “clot busters” are given within a few hours of the infarction.
An acute interruption in coronary blood flow is most often a result of the sudden arrival of a blood clot (eg, an embolism released from a clot in a fibrillating atrium) or the formation of an intravascular clot at the site of a ruptured atherosclerotic plaque. Either of these events may abruptly occlude or significantly narrow a major coronary artery. This is indeed a crisis situation and demands immediate attention. The physiological consequences of such an immediate occlusion are discussed in the preceding text under the topic of “Cardiogenic Shock.” Treatments may include emergency coronary angioplasty (described earlier) with the placement of a stent, which may allow immediate restoration of flow to the ischemic area. Another method for treatment of acute myocardial infarction is the intravascular injection of thrombolytic substances (eg, streptokinase or tissue plasminogen-activating factors) that dissolve blood clots. This approach is most successful when these “clot busters” are given within a few hours of the infarction.
Chronic Heart Failure—Systolic Dysfunction
![]() Heart (or cardiac or myocardial) failure is said to exist whenever ventricular function is depressed through myocardial damage, insufficient coronary flow, or any other condition that directly impairs the mechanical performance of the heart muscle. By definition, systolic heart failure is associated with a left ventricular ejection fraction of less than 40%. This also implies that the heart is operating on a lower-than-normal cardiac function curve, that is, a reduced cardiac output at any given filling pressure. Acute heart failure has already been discussed in the preceding text in the context of sudden coronary artery occlusion, cardiogenic shock, and as part of the decompensatory mechanisms operating in progressive and irreversible shock. Often, however, sustained cardiac “challenges” may induce a chronic state of heart failure. Such challenges might include (1) progressive coronary artery disease, (2) sustained elevation in cardiac afterload as that which accompanies arterial hypertension or aortic valve stenosis, or (3) reduced functional muscle mass following myocardial infarction. In some instances, external causes of cardiac failure cannot be identified and some primary genetic abnormality in myocyte cytoskeletal or sarcomeric protein is to blame. This situation is referred to as primary cardiomyopathy. Regardless of the precipitating cause, most forms of failure are associated eventually with a reduced myocyte function. Many specific structural, functional, and biochemical myocyte alterations accompany severe systolic heart failure. Some of the more well-documented abnormalities include (1) reduced calcium sequestration by the sarcoplasmic reticulum and upregulation of the sarcolemmal Na/Ca exchanger (leading to low intracellular calcium levels for excitation–contraction coupling), (2) low affinity of troponin for calcium (leading to reduced cross-bridge formation and contractile ability), (3) altered substrate metabolism from fatty acid to glucose oxidation, and (4) impaired respiratory chain activity (leading to impaired energy production).
Heart (or cardiac or myocardial) failure is said to exist whenever ventricular function is depressed through myocardial damage, insufficient coronary flow, or any other condition that directly impairs the mechanical performance of the heart muscle. By definition, systolic heart failure is associated with a left ventricular ejection fraction of less than 40%. This also implies that the heart is operating on a lower-than-normal cardiac function curve, that is, a reduced cardiac output at any given filling pressure. Acute heart failure has already been discussed in the preceding text in the context of sudden coronary artery occlusion, cardiogenic shock, and as part of the decompensatory mechanisms operating in progressive and irreversible shock. Often, however, sustained cardiac “challenges” may induce a chronic state of heart failure. Such challenges might include (1) progressive coronary artery disease, (2) sustained elevation in cardiac afterload as that which accompanies arterial hypertension or aortic valve stenosis, or (3) reduced functional muscle mass following myocardial infarction. In some instances, external causes of cardiac failure cannot be identified and some primary genetic abnormality in myocyte cytoskeletal or sarcomeric protein is to blame. This situation is referred to as primary cardiomyopathy. Regardless of the precipitating cause, most forms of failure are associated eventually with a reduced myocyte function. Many specific structural, functional, and biochemical myocyte alterations accompany severe systolic heart failure. Some of the more well-documented abnormalities include (1) reduced calcium sequestration by the sarcoplasmic reticulum and upregulation of the sarcolemmal Na/Ca exchanger (leading to low intracellular calcium levels for excitation–contraction coupling), (2) low affinity of troponin for calcium (leading to reduced cross-bridge formation and contractile ability), (3) altered substrate metabolism from fatty acid to glucose oxidation, and (4) impaired respiratory chain activity (leading to impaired energy production).
The primary disturbance in systolic heart failure (acute or chronic) is depressed cardiac output and thus lowered arterial pressure. Consequently, all the compensatory responses important in shock (Figure 11–1) are also important in heart failure. In chronic heart failure, however, the cardiovascular disturbances may not be sufficient to produce a state of shock. Moreover, long-term compensatory mechanisms are especially important in chronic heart failure.
The circumstances of chronic systolic heart failure are well illustrated by cardiac output and venous function curves such as those shown in Figure 11–3. The normal cardiac output and normal venous function curves intersect at point A in Figure 11–3. A cardiac output of 5 L/min at a central venous pressure of less than 2 mm Hg is indicated by the normal operating point (A). With heart failure, the heart operates on a much lower-than-normal cardiac output curve. Thus, acute heart failure alone (uncompensated) shifts the cardiovascular operation from the normal point (A) to a new position, as illustrated by point B in Figure 11–3—that is, cardiac output falls below normal while central venous pressure rises above normal. The decreased cardiac output leads to decreased arterial pressure and reflex activation of the cardiovascular sympathetic nerves. Increased sympathetic nerve activity tends to (1) raise the cardiac function curve toward normal and (2) increase peripheral venous pressure through venous constriction, and thus raise the venous function curve above normal. Cardiovascular operation will shift from point B to point C in Figure 11–3. Thus, the depressed cardiac output is substantially improved by the immediate consequences of increased sympathetic nerve activity. Note, however, that the cardiac output at point C is still below normal. The arterial pressure associated with cardiovascular operation at point C is likely to be near normal, however, because higher-than-normal total peripheral resistance will accompany higher-than-normal sympathetic nerve activity.
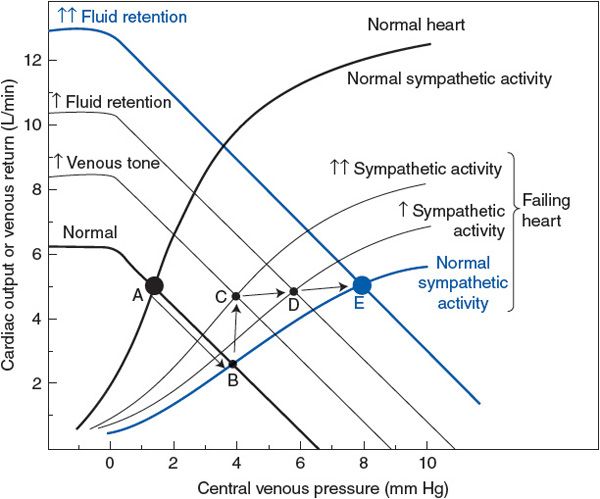
Figure 11–3. Cardiovascular alterations with compensated chronic systolic heart failure.
![]() In the long term, cardiovascular operation cannot remain at point C in Figure 11–3. Operation at point C involves higher-than-normal sympathetic activity, and this will inevitably cause a gradual increase in blood volume by the mechanisms that are described in Chapter 9. Over several days, there is a progressive rise in the venous function curve as a result of increased blood volume and, consequently, increased mean circulatory filling pressure. Recall that this process involves a sympathetically induced release of renin from the kidney, which activates the renin–angiotensin–aldosterone system that promotes fluid retention. This will progressively shift the cardiovascular operating point from C to D to E, as shown in Figure 11–3.
In the long term, cardiovascular operation cannot remain at point C in Figure 11–3. Operation at point C involves higher-than-normal sympathetic activity, and this will inevitably cause a gradual increase in blood volume by the mechanisms that are described in Chapter 9. Over several days, there is a progressive rise in the venous function curve as a result of increased blood volume and, consequently, increased mean circulatory filling pressure. Recall that this process involves a sympathetically induced release of renin from the kidney, which activates the renin–angiotensin–aldosterone system that promotes fluid retention. This will progressively shift the cardiovascular operating point from C to D to E, as shown in Figure 11–3.
Note that increased fluid retention (C → D → E in Figure 11–3) causes a progressive increase in cardiac output toward normal and simultaneously allows a reduction in sympathetic nerve activity toward the normal value. Reduced sympathetic activity is beneficial for several reasons. First, decreased arteriolar constriction permits renal and splanchnic blood flow to return toward more normal values. Second, myocardial oxygen consumption may fall as sympathetic nerve activity falls, even though cardiac output tends to increase. Recall that the increased heart rate and increased cardiac contractility caused by sympathetic nerve activation greatly increase myocardial oxygen consumption. Reduced myocardial oxygen consumption is especially beneficial in situations where inadequate coronary blood flow is the cause of the heart failure. In any case, once enough fluid has been retained that a normal cardiac output can been achieved with normal sympathetic nerve activity, the individual is said to be in a “compensated” state of chronic heart failure.3
Unfortunately, the consequences of fluid retention in chronic cardiac failure are not all beneficial. Note that in Figure 11–3 fluid retention (C → D → E) will cause both peripheral and central venous pressures to be much higher than their normal values. Chronically high central venous pressure causes chronically increased end-diastolic volume (cardiac dilation). Up to a point, cardiac performance is very much improved by increased cardiac filling volume. Excessive cardiac dilation, however, can impair cardiac function because increased total wall tension is required to generate pressure within an enlarged ventricular chamber (T = P × r; Chapter 2). This increases the myocardial oxygen demand.
The high venous pressure associated with fluid retention also adversely affects organ function because high venous pressure produces transcapillary fluid filtration, edema formation, and congestion (hence the commonly used term congestive heart failure). Left-sided heart failure is accompanied by pulmonary edema with dyspnea (shortness of breath) and respiratory crisis.4 Right-sided heart failure is associated with distended neck veins, ankle edema, and fluid accumulation in the abdomen (ascites) with liver congestion and dysfunction.5
In the example shown in Figure 11–3, the depression in the cardiac output curve because of heart failure is only moderately severe. Thus, it is possible, through moderate fluid retention, to achieve a normal cardiac output with essentially normal sympathetic activity (point E). The situation at point E is relatively stable because the stimuli for further fluid retention have been removed. If, however, the heart failure is more severe, the cardiac output curve may be so depressed that normal cardiac output cannot be achieved by any amount of fluid retention. In these cases fluid retention is extremely marked, as is the elevation in venous pressure, and the complications of congestion are very serious problems.
Another way of looking at the effects of left ventricular cardiac failure is given in Figure 11–4. The left ventricular pressure–volume loops describing the events of a cardiac cycle from a failing heart are displaced far to the right of those from normal hearts. The untreated patient described in this figure is in serious trouble with a reduced stroke volume and ejection fraction and high filling pressure. Furthermore, the slope of the line describing the end-systolic pressure–volume relationship is shifted downward and is less steep, indicating the reduced contractility of the cardiac muscle. However, because of this flatter relationship, small reductions in cardiac afterload (ie, arterial blood pressure) will produce substantial increases in stroke volume, as indicated in Figure 11–4, and will significantly help this patient.
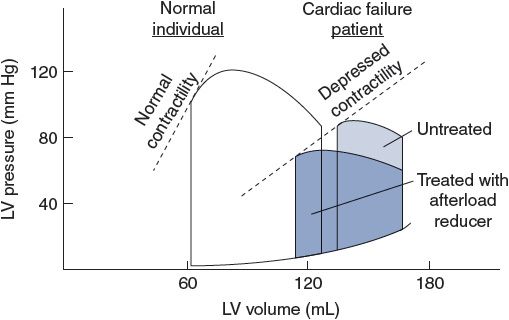
Figure 11–4. Left ventricular pressure–volume loops showing systolic heart failure (loops to the right) characterized by depressed contractility, increased end-diastolic volume, and reduced ejection fraction.
As might be expected from the previous discussion, the most common symptoms of patients with congestive heart failure are associated with the inability to increase cardiac output (low exercise tolerance and fatigue) and with the compensatory fluid accumulation (tissue congestion, shortness of breath, and peripheral swelling). In severe cases, the ability of the cardiac cells to respond to increases in sympathetic stimulation is diminished by a reduction in the effective number (downregulation) of the myocyte β1-adrenergic receptors. This further reduces the ability of the myocytes to increase their contractility as well as the ability of the heart to increase its beating rate in response to sympathetic stimulation. Thus, low maximal heart rates contribute to the reduced exercise tolerance.
Treatment of the patient with congestive systolic heart failure is a difficult challenge. Treatment of the precipitating condition is of course the ideal approach, but often this cannot be done effectively. Cardiac glycosides (eg, digitalis)6 have been used to improve cardiac contractility (ie, to increase the ejection fraction, shift the cardiac function curve upward, increasing contractile force of the myocyte at any given starting length).7 These drugs are unfortunately quite toxic and often have undesirable side effects.
Treatment of the congestive symptoms involves balancing the need for enhanced cardiac filling with the problems of too much fluid. Drugs that promote renal fluid loss (diuretics such as furosemide or thiazides) are extremely helpful as are the angiotensin-converting enzyme (ACE) inhibitors and the angiotensin II receptor blockers (ARBs).8 A potent diuretic can quickly save a patient from drowning in the pulmonary exudate and reduce diastolic volume of the dilated heart to acceptable levels, but it can also lower blood pressure to dangerous levels.
Chronic heart failure patients often have elevated sympathetic drive if they have not completely compensated for the depressed cardiac function by fluid retention and increased blood volume. Although high sympathetic drive is an important initial compensatory mechanism, the energy cost of a chronically elevated heart rate and contractility can put the heart with diminished coronary circulation at a disadvantage. Therefore, treatment with β-adrenergic receptor blockers can reduce metabolic demand to a level more easily met by a compromised vascular supply. Again, if elimination of the effect of elevated sympathetic drive on the heart is too aggressive, cardiac output may fall and worsen a failure state.
Chronic Heart Failure—Diastolic Dysfunction
![]() Although systolic dysfunction is often the primary cause of heart failure, some degree of diastolic dysfunction is also commonly present and may precede systolic problems. As shown in Figure 11–5, diastolic dysfunction implies a stiffened heart during diastole such that increases in cardiac filling pressure do not produce normal increases in end-diastolic volume. Some individuals (primarily elderly patients with hypertension and cardiac hypertrophy) who have some symptoms of cardiac failure (exertional dyspnea, fluid retention, pulmonary edema, and high end-diastolic pressures) seem to have normal systolic function (ejection fractions >40%), and normal or even reduced ventricular end-diastolic volumes despite increased cardiac filling pressure. Thus, the terms diastolic heart failure and heart failure with preserved systolic function have been used to describe this situation.
Although systolic dysfunction is often the primary cause of heart failure, some degree of diastolic dysfunction is also commonly present and may precede systolic problems. As shown in Figure 11–5, diastolic dysfunction implies a stiffened heart during diastole such that increases in cardiac filling pressure do not produce normal increases in end-diastolic volume. Some individuals (primarily elderly patients with hypertension and cardiac hypertrophy) who have some symptoms of cardiac failure (exertional dyspnea, fluid retention, pulmonary edema, and high end-diastolic pressures) seem to have normal systolic function (ejection fractions >40%), and normal or even reduced ventricular end-diastolic volumes despite increased cardiac filling pressure. Thus, the terms diastolic heart failure and heart failure with preserved systolic function have been used to describe this situation.
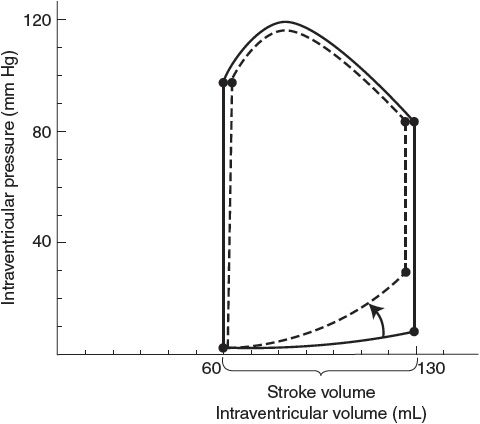
Figure 11–5. Left ventricular pressure–volume loops showing diastolic heart failure (dashed lines) characterized by increased diastolic stiffness, increased end-diastolic pressure, and normal ejection fraction.
Potential causes of altered diastolic properties in heart failure include (1) decreased cardiac tissue passive compliance due to extracellular remodeling, collagen cross-linking, and other extracellular matrix protein alterations, (2) increased myofibrillar passive stiffness due to alterations in the myofibrillar protein titin, (3) delayed myocyte relaxation early in diastole due to slow cytosolic calcium removal processes, (4) inadequate adenosine triphosphate levels required to disconnect the myofilament cross-bridges rapidly, and (5) residual, low-grade cross-bridge cycling during diastole due to calcium leaking from the sarcoplasmic reticulum.
At this point, therapeutic strategies that directly influence diastolic properties are not well developed. Attempts to reduce interstitial fibrosis (with ACE inhibitors and/or angiotensin receptor antagonists) and to reduce diastolic calcium leak from the sarcoplasmic reticulum (with β-adrenergic blockers) have had limited success. Reduction of afterload seems to be most helpful, especially if it reduces left ventricular hypertrophy.
HYPERTENSION
Hypertension is defined as a chronic elevation in arterial blood pressure and can exist in either the pulmonary or the systemic vascular system.
![]() Pulmonary hypertension is much less common than systemic hypertension and less is known about its causes, progression, and treatments. Pulmonary hypertension is designated when mean pulmonary artery pressure is greater than 20 mm Hg. It is relatively hard to diagnose until the consequences of the elevated pulmonary arterial pressures (systemic edema, chest pain, and fatigue) are fairly well advanced. Right-sided heart failure resulting from chronic pulmonary hypertension is called cor pulmonale. Although there is a genetic component to its incidence, it seems to be highly correlated with conditions involving chronic hypoxia (eg, chronic obstructive pulmonary disease, cystic fibrosis, and pneumoconiosis).9 The increased prevalence of obesity with accompanying obstructive sleep apnea may account for a recent increase in reported incidence of this disease. There is no cure at present and strategies for treatment of systemic hypertension have little or no effect on pulmonary hypertension. However, three pharmacological approaches are useful: (1) endothelin receptor antagonists block the biological activities of the vasoconstrictor, endothelin; (2) phosphodiesterase-5 inhibitors prevent breakdown of cyclic guanosine monophosphate, thus promoting the vasorelaxing effects of nitric oxide; and (3) prostacyclin derivatives provide an exogenous supply of the vasodilator, prostacyclin.
Pulmonary hypertension is much less common than systemic hypertension and less is known about its causes, progression, and treatments. Pulmonary hypertension is designated when mean pulmonary artery pressure is greater than 20 mm Hg. It is relatively hard to diagnose until the consequences of the elevated pulmonary arterial pressures (systemic edema, chest pain, and fatigue) are fairly well advanced. Right-sided heart failure resulting from chronic pulmonary hypertension is called cor pulmonale. Although there is a genetic component to its incidence, it seems to be highly correlated with conditions involving chronic hypoxia (eg, chronic obstructive pulmonary disease, cystic fibrosis, and pneumoconiosis).9 The increased prevalence of obesity with accompanying obstructive sleep apnea may account for a recent increase in reported incidence of this disease. There is no cure at present and strategies for treatment of systemic hypertension have little or no effect on pulmonary hypertension. However, three pharmacological approaches are useful: (1) endothelin receptor antagonists block the biological activities of the vasoconstrictor, endothelin; (2) phosphodiesterase-5 inhibitors prevent breakdown of cyclic guanosine monophosphate, thus promoting the vasorelaxing effects of nitric oxide; and (3) prostacyclin derivatives provide an exogenous supply of the vasodilator, prostacyclin.
![]() Systemic hypertension is defined as an elevation of mean systemic arterial pressure above 140/90 mm Hg. It is an extremely common cardiovascular problem, affecting more than 20% of the adult population of the western world. It has been established beyond doubt that hypertension increases the risk of coronary artery disease, myocardial infarction, heart failure, stroke, and many other serious cardiovascular problems. Moreover, it has been clearly demonstrated that the risk of serious cardiovascular incidents is reduced by proper treatment of hypertension.10
Systemic hypertension is defined as an elevation of mean systemic arterial pressure above 140/90 mm Hg. It is an extremely common cardiovascular problem, affecting more than 20% of the adult population of the western world. It has been established beyond doubt that hypertension increases the risk of coronary artery disease, myocardial infarction, heart failure, stroke, and many other serious cardiovascular problems. Moreover, it has been clearly demonstrated that the risk of serious cardiovascular incidents is reduced by proper treatment of hypertension.10
In approximately 90% of cases, the primary abnormality that produces high blood pressure is unknown. This condition is sometimes referred to as primary or essential hypertension because the elevated level was thought to be “essential” to drive the blood through the systemic circulation. In the remaining 10% of hypertensive patients, the cause can be traced to a variety of sources, including epinephrine-producing tumors (pheochromocytomas), aldosterone-producing tumors (in primary hyperaldosteronism), certain forms of renal disease (eg, renal artery stenosis, glomerular nephritis, and toxemia of pregnancy), certain neurological disorders (eg, brain tumors that increase intracranial pressure), certain thyroid and parathyroid disorders, aortic coarctation, lead poisoning, drug side effects, abuse of certain drugs, obstructive sleep apnea, or even unusual dietary habits. The high blood pressure that accompanies such known causes is referred to as secondary hypertension. Most often, however, the true cause of the hypertension remains a mystery, and it is only the symptom of high blood pressure that is treated.
Facts about Systemic Hypertension
In the midst of an enormous amount of information about systemic hypertension, a few universally accepted facts stand out:
1. Genetic factors contribute importantly to the development of hypertension. Familial tendencies for high blood pressure are well documented. In addition, hypertension is generally more common in men than in women and in blacks than in whites.
2. Chronic conditions such as obesity, diabetes, kidney disease, and sleep disorders are strongly associated with systemic hypertension.
3. Environmental factors or behaviors can influence the development of hypertension. Physical inactivity, use of tobacco, excess alcohol consumption, high-sodium or low-potassium diets, and/or certain forms of psychological stress may either aggravate or precipitate hypertension in genetically susceptible individuals.
4. Structural changes in the left side of the heart and arterial vessels occur in response to systemic hypertension. Early alterations include hypertrophy of muscle cells and thickening of the walls of the ventricle and systemic resistance vessels. Late changes associated with deterioration of function include increases in connective tissue and loss of elasticity.
5. The established phase of hypertension is associated with an increase in total peripheral resistance. Cardiac output and/or blood volume may be elevated during the early developmental phase, but these variables are usually normal after the hypertension is established.
6. The increased total peripheral resistance associated with established hypertension may be due to (a) rarefaction (decrease in density) of microvessels, (b) pronounced structural adaptations that occur in the peripheral vascular bed, (c) continuously increased activity of the vascular smooth muscle cells,11 (d) increased sensitivity and reactivity of the vascular smooth muscle cells to external vasoconstrictor stimuli, and/or (e) diminished production and/or effect of endogenous vasodilator substances (eg, nitric oxide).
7. The chronic elevation in blood pressure does not appear to be due to a sustained elevation in sympathetic vasoconstrictor neural discharge nor is it due to a sustained elevation of any blood-borne vasoconstrictive factor. (Both neural and hormonal influences, however, may help initiate primary hypertension.)
8. Blood pressure–regulating reflexes (both the short-term arterial and cardiopulmonary baroreceptor reflexes and the long-term, renal-dependent, pressure-regulating reflexes) become adapted or “reset” to regulate blood pressure at a higher-than-normal level.
9. Disturbances in renal function contribute importantly to the development and maintenance of primary hypertension. Recall that the urinary output rate is influenced by arterial pressure, and, in the long term, arterial pressure can stabilize only at the level that makes urinary output rate equal to fluid intake rate. As shown by point N in Figure 11–6, this pressure is approximately 100 mm Hg in a normal individual.
All forms of hypertension involve an alteration somewhere in the chain of events by which changes in arterial pressure produce changes in urinary output rate (see Figure 9–6) such that the renal function curve is shifted rightward, as indicated in Figure 11–6. The important feature to note is that higher-than-normal arterial pressure is required to produce a normal urinary output rate in a hypertensive individual. Although this condition is always present with hypertension, it is not clear whether it could be the common cause of hypertension or simply another one of the many adaptations to it.
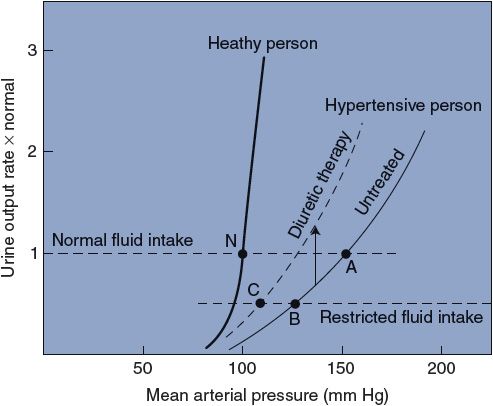
Figure 11–6. Renal function curves in healthy and hypertensive people.
Consider that the untreated hypertensive individual in Figure 11–6 would have a very low urinary output rate at the normal mean arterial pressure of 100 mm Hg. Recall from Figure 9–5 that whenever the fluid intake rate exceeds the urinary output rate, fluid volume must rise and consequently so will cardiac output and mean arterial pressure. With a normal fluid intake rate, this untreated hypertensive patient will ultimately stabilize at point A (mean arterial pressure 150 mm Hg). Recall from Chapter 9 that the baroreceptors adapt within days so that they have a normal discharge rate at the prevailing average arterial pressure. Thus, once the hypertensive individual has been at point A for a week or more, even the baroreceptor mechanism will begin resisting acute changes from the 150-mm Hg pressure level.
A most important fact to realize is that, although either high cardiac output or high total peripheral resistance must always ultimately sustain high blood pressure, neither needs be the primary cause of the hypertension. A shift in the relationship between arterial pressure and urinary output rate, as illustrated in Figure 11–6, however, will always produce hypertension. The possibility that the kidneys actually “set” the blood pressure is supported by evidence accumulating from kidney transplant studies. In these studies, the blood pressure is shown to “follow” the kidney (ie, putting a hypertensive kidney in a normotensive individual produces a hypertensive individual, whereas putting a normotensive kidney in a hypertensive individual produces a normotensive individual). Further support for an essential role of the kidney comes from recent studies showing that catheter-based, high-frequency radiowave ablation of renal sympathetic nerves effectively reduces hypertension in drug-resistant patients.
Therapeutic Strategies for Treatment of Systemic Hypertension
In certain hypertensive individuals, restricting salt intake produces a substantial reduction in blood pressure because of the reduced requirement for water retention to osmotically balance the salt load. In the example in Figure 11–6, this effect is illustrated by a shift from point A to point B. The efficacy of lowering salt intake to lower arterial pressure depends heavily on the slope of the renal function curve in the hypertensive individual. The arterial pressure of a healthy individual, for example, is affected only slightly by changes in salt intake because the normal renal function curve is so steep.
A second common treatment of hypertension is diuretic therapy. Many diuretic drugs are available, but most have the effect of inhibiting renal tubular salt (and therefore fluid) reabsorption. The net effect of diuretic therapy, as shown in Figure 11–6, is that the urinary output rate for a given arterial pressure is increased; that is, diuretic therapy raises the renal function curve. The combined result of restricted fluid intake and diuretic therapy for the hypertensive individual in Figure 11–6 is illustrated by point C.
A third therapeutic intervention is treatment with β-adrenergic blockers that inhibit sympathetic influences on the heart and renal renin release. This approach is most successful in hypertensive patients who have high circulating renin levels.
A fourth antihypertensive strategy is to block the effects of the renin–angiotensin system either with ACE inhibitors blocking the formation of the vasoconstrictor angiotensin II or with angiotensin II receptor blockers. Other pharmacological interventions may include use of α-adrenergic receptor blockers, which prevent the vasoconstrictive effects of catecholamines, and calcium channel blockers, which act directly to decrease vascular smooth muscle tone.
Alterations in life style, including reduction of stress, decreases in caloric intake, limitation of the amount of saturated fats in the diet, and establishment of a regular exercise program, may help reduce blood pressure in certain individuals.
The possibility of catheter-based renal sympathetic denervation for treatment of refractory hypertension is quite intriguing and may become another approach to handling this condition in the future.
PERSPECTIVES
Too often, students (and some physicians) learn to recognize a list of symptoms and leap to a diagnosis based upon empirical approaches. Treatment may be based upon a dictated “standard of care” and, although concern for the patient is present, the thought process is limited. We hope that this chapter’s overview of common abnormalities in the cardiovascular system will reassure the student that memorizing everything is not necessary. Understanding the basic principles of normal cardiovascular operation should provide a firm foundation for identifying the underlying abnormalities, distinguishing the primary disturbances from the compensatory responses, understanding the mechanisms responsible for the symptoms, and appropriately treating the condition.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


