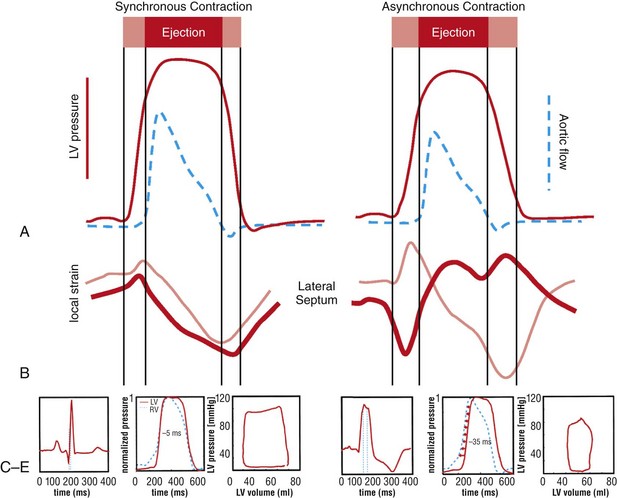
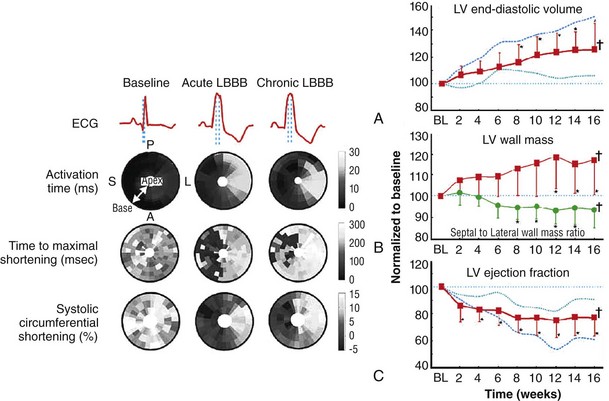
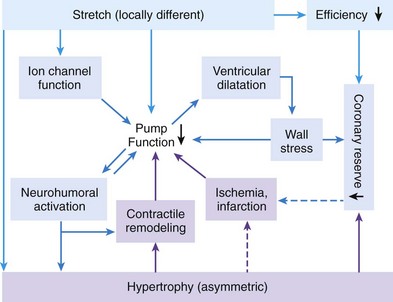
118 Electromechanical Events and Cardiac Pump Function in Asynchronous Heart Failure Consequences of Asynchrony at Various Levels Mechanisms of Cardiac Resynchronization Therapy (CRT) Programming Considerations for CRT Tailoring CRT Programming to Improve Cardiac Mechanics and to Maximize the Probability of Reverse Volumetric LV Remodeling Normal LV electrical activation is rapid and homogeneous with minimal temporal dispersion. This elicits synchronous mechanical activation and ventricular contraction. The resulting coordinated myocardial segment activation maximizes ventricular pump function. Wiggers established the linkage between pacing-induced alterations in ventricular conduction and pump function with two seminal observations in 1925: (1) Stimulation at virtually any ventricular site disturbs the natural pattern of activation and contraction because the evoked electrical wavefront propagates slowly through ventricular myocardium rather than through the Purkinje system; and (2) altered ventricular activation causes an immediate reduction in pump function.1 Delayed intraventricular activation is the most important determinant of reduced pump function in LBBB. Electrical activation starts in the interventricular septum, and the posterolateral LV wall is activated >100 ms later. Such considerable intraventricular delay during LBBB is due to slow spread of the depolarization wavefront through the working myocardium rather than through the Purkinje system. LBBB is a complex electrical disease that results from LV conduction delay at multiple anatomical levels; it may be anatomically fixed or functional and is exhibited differently according to substrate characterization (ischemic vs. nonischemic cardiomyopathy). High-resolution endocardial and epicardial mapping studies have revealed that LV activation during LBBB, despite a similar surface electrocardiographic appearance, is heterogeneous. At least three patterns of delayed LV activation have been characterized: (1) transseptal delay with or without LV endocardial delay, (2) normal transseptal conduction with slow conduction velocities in peri-infarct regions or globally slow in nonischemic disease, and (3) slow U-shaped activation around a line of functional block on the anterior wall.2–4 Generally, QRS duration (QRSd) of 120 to 150 ms indicates delay confined specifically to the specialized conduction system, whereas QRSd >150 ms usually indicates additional conduction delay in diseased myocardium. Nominally, the posterolateral basal LV is the latest activated region. The pattern of LV activation is also influenced by the location and size of lines of fixed or functional conduction block. Fixed conduction block is due to replacement of normal myocardium by scar. The physiological basis for functional conduction blocks has not been elucidated but could relate to stretch, heart rate, and spontaneous diastolic depolarization. Anterior locations of the line of functional block are characterized by a U-shaped LV activation pattern,3 more prolonged QRSd (>150 ms), and greater time to LV breakthrough.3 Late activation of the posterolateral basal LV occurred by wavefront propagation around the line of block using the apical or inferior LV walls. Lateral locations of the lines of block are characterized by less prolonged QRSd (<150 ms) and shorter time to LV breakthrough. Pacing maneuvers shift either line of block, indicating their functional nature.3 Noninvasive mapping of epicardial activation using body surface potentials demonstrates that electrical activation patterns in LBBB are highly heterogeneous and unpredictable.4 Lines of conduction block do not correlate with regions of wall motion abnormality or scarring. Some lines of block arose only during pacing and were site dependent (functional), whereas other lines of block could not be manipulated with pacing maneuvers, indicating that these were due to slow or absent conduction (fixed). Latest activation most often occurred in the posterolateral wall but was also observed in the anterior and inferior walls in some patients. Regions that are activated early also start to contract early. In LBBB earliest and latest sites of segmental LV activation correlate well with time to peak systolic velocity by Doppler and strain by tagged magnetic resonance imaging (MRI), providing evidence that ventricular conduction delay is responsible for mechanical asynchrony. The effect is dramatic because the various regions differ not only in the time of onset of contraction but also in the pattern of contraction (Figure 118-1). Contraction of early-activated myocardium is energetically inefficient because LV pressure is low and ejection has not begun. Instead, stretching of not as yet activated remote regions absorbs the energy generated by the early-activated regions. This stretching further delays shortening of these late activation regions and increases their force of local contraction by the Frank-Starling mechanism (locally enhanced preload). Vigorous late systolic contraction at delayed sites occurs against high LV cavity pressures (locally enhanced afterload) and imposes loading on the earlier-activated regions, which undergo systolic paradoxical stretch. This reciprocated stretching of regions within the LV wall causes a less effective and energetically efficient contraction. Figure 118-1 Effects of synchronous (left panel) and asynchronous (right panel) ventricular activation on LV and aortic pressure (A), regional strain (B), ventricular conduction (C, electrocardiographic tracings), (D) RV and LV pressure signals, and (E) pressure-volume loops. Asynchronous contraction causes reduction in ejection time and slows rates of rise and fall of LV pressure and aortic pressure and increases the duration of isovolumetic contraction (ic) and relaxation (ir) (A). Onset of LV shortening (strain) is regionally delayed (negative deflection of curve) (B). Asynchronous electromechanical activation induces increased QRSd (C) and interventricular/intraventricular activation times (D), which instantaneously reduce stroke volume, stroke work, dP/dt max, and dP/dt min, indicating an acute reduction in pump function (E). (Adapted from Sweeney MO, Prinzen FW: Ventricular pump function and pacing: Physiological and clinical integration. Circ Arrhythm Electrophysiol 1:127-139, 2008.) Hemodynamic consequences of the asynchronous LV contraction include reductions in contractility and relaxation. These changes occur immediately upon induction of LBBB.5 Loss of pump function is indicated by decreases in stroke volume and stroke work and slower rates of rise of LV pressure (Figure 118-2). Moreover, the LV end-systolic pressure-volume relationship shifts rightward, indicating that the LV operates at a larger volume to recruit the Frank-Starling mechanism. Premature relaxation in early-activated regions and delayed contraction in others also cause abnormal relaxation, expressed as a slower rate of fall of LV pressure. These changes lead to prolongation of isovolumetic contraction and relaxation times, which is characteristic of asynchronous hearts. Prolongation of isovolumetic times occurs mainly at the expense of diastolic filling time, leading to reduced preload. Figure 118-2 LBBB-Induced Asynchrony Causes Immediate Reduction in LV Contractility Redistribution of the mechanical load within the ventricular wall also leads to reduction of regional myocardial perfusion and oxygen consumption in the septum.6 These defects express regional differences in myocardial workload, are reversible, and are noted after biventricular pacing.6 Chronic asynchronous LV activation results in regional and global structural changes indicated by asymmetric hypertrophy, increased end-diastolic volume, and reduced ejection fraction (Figure 118-3),6 as well as locally different molecular abnormalities including reductions in sarcoplasmic reticulum calcium–adenosine triphosphatase (ATPase) and phospholamban. Figure 118-3 Left, Effects of induced LBBB on electromechanics. Dashed lines on surface ECG indicate earliest and latest LV endocardial activation times. Bull’s eye plots indicate septal (S), posterior (P), lateral (L), and anterior (A) LV wall, where the outer ring indicates the LV base, and the inner ring indicates the LV apex. Activation is color coded; lighter shades indicate later activation times. During normal ventricular conduction (Baseline), QRSd is narrow and LV electromechanical activation is simultaneous throughout. During LBBB, QRS is prolonged and LV electrical and mechanical activation is delayed with lateral wall latest. Right, Change in LV end-diastolic volume (increase), LV septal-lateral wall mass ratio (decrease, indicating asymmetric hypertrophy), and ejection fraction (decrease) after 16 weeks of CRT. (Modified from Sweeney MO, Prinzen FW: Ventricular pump function and pacing: Physiological and clinical integration. Circ Arrhythm Electrophysiol 1:127-139, 2008.) Similar to the situation after myocardial infarction, acute loss of pump function initiates compensatory responses (Figure 118-4). Some of these responses, after a certain time or a certain degree of asynchrony, may lead to further impairment of pump function and clinical heart failure. Various triggers for these “remodeling” processes have been identified. As is the case for other conditions of hemodynamic overload, LBBB leads to stimulation of the sympathetic system, resulting in elevated myocardial catecholamine levels and activation of the renin-angiotensin-aldosterone system. Regional differences in stretch and mechanical load heterogeneity are most likely important stimuli for remodeling processes. Figure 118-4 Mechanisms of ventricular remodeling and progressive reduction in pump function during mechanical dys-synchrony induced by ventricular conduction delay (LBBB). For details, see text. (Adapted from Sweeney MO, Prinzen FW: Ventricular pump function and pacing: Physiological and clinical integration. Circ Arrhythm Electrophysiol 1:127-139, 2008.)
Cardiac Resynchronization Therapy
Consequences of Asynchrony at Various Levels
Ventricular Asynchrony
Effects of Asynchrony on LV Mechanics and Structure
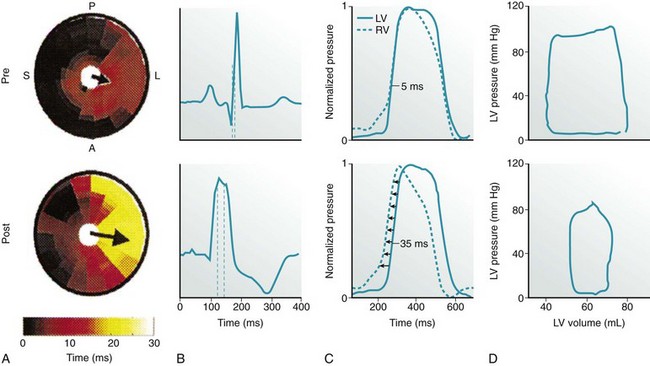
A, LV endocardial activation maps. Inner circle represents LV apex, and outer circle represents LV base. S, A, L, and P indicate the septum and anterior, lateral, and posterior walls, respectively. Arrows indicate activation delay vectors, the amplitude of which reflects the degree of intraventricular asynchrony (intraVA). B, Surface ECG tracings, where QRS duration indicates total ventricular asynchrony (total VA). C, RV and LV pressure signals normalized to reveal pressure differences (interventricular asynchrony, interVA). D, Pressure-volume (PV) loops. Loop area is stroke work; loop width is stroke volume.
During normal ventricular conduction (top row, pre-LBBB), intraVA and interVA are minimized, QRSd is normal, and LV contractility is greater (larger PV loop). After induction of LBBB (bottom row, post-LBBB), intraVA, interVA, and total VA are increased; this immediately reduces LV pump function (smaller PV loop indicating a decline in stroke work, stroke volume, and LV pressure generation). (Adapted from Sweeney MO, Prinzen FW: Ventricular pump function and pacing: Physiological and clinical integration. Circ Arrhythm Electrophysiol 1:127-139, 2008.)
Thoracic Key
Fastest Thoracic Insight Engine



