By location
By receptor types
Cardiac receptors
• Myocardial
• Conduction system
• Others
G protein-coupled receptors
Tyrosine kinase-linked receptors
Guanylyl cyclase-linked receptors
Vascular receptors
• Endothelial
• Vascular smooth muscle
• Others
Other receptors (low-sensitivity lipoprotein receptor; nicotine acetylcholine receptor; peptidergic)
15.3 G Protein-Coupled Receptor (Seven-Transmembrane-Spanning Receptors) and Signal Transduction
15.3.1 Overview
G protein-coupled receptors are functionally closely related to ion channels. The G protein-coupled receptors are part of a gene family being identified that binds agonists such as adenosine, catecholamines, acetylcholine, odorants, angiotensin, histamine, opioids, and many others.
15.3.2 Receptor Structure
There are several interesting structural similarities between ion channels and G protein-coupled receptors. Both are integral membrane proteins with seven-transmembrane domains, which form bundles with a central pocket. In G proteins, the pockets are the binding sites for the receptor ligands, and they are typically located on the extracellular site of the protein; the N-terminal tail is located extracellularly as are three extracellular loops. Three loops connecting the transmembrane domains and the C-terminal domain are located intracellularly (Figs. 15.1 and 15.2). The exact function of the extracellular loops remains largely unknown. It is considered that the transmembrane domains are involved in receptor binding. The intracellular loops, particularly loop III and the C-terminal tail, are important for receptor coupling to the associated G protein. Finally, both loop III and C-terminal tail are important for regulation of receptor function and contain phosphorylation and other posttranslational modification sites .
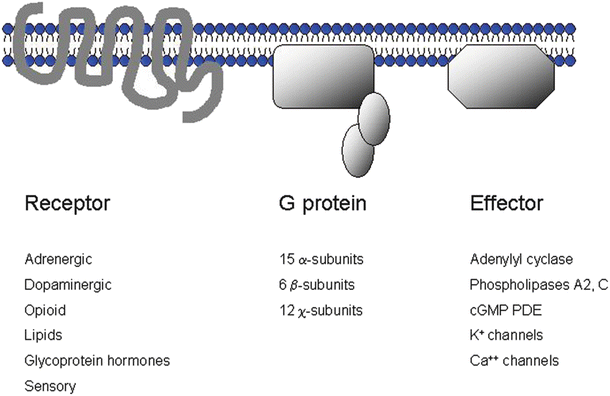
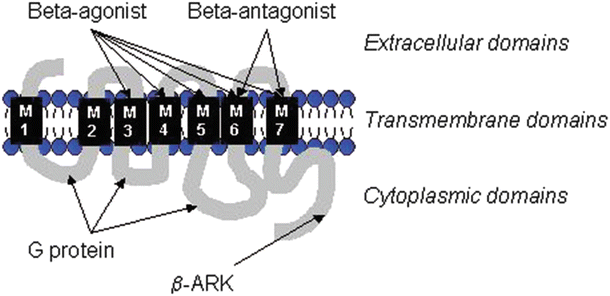
Fig. 15.1
G protein receptor-coupled signaling. The ubiquitous seven-transmembrane receptor systems are composed of a receptor, a heterotrimeric G protein, and an effector system. The complexity of this system can be appreciated by the number of receptor types and subtypes, G protein subtypes, and different effector systems
Fig. 15.2
The molecular schematic structure of a β-adrenergic receptor. Note the three main domains—extracellular, transmembrane, and cytoplasmic. The transmembrane domains are important for ligand binding. The transmembrane domain, M3-7, is important in agonist binding, whereas the domains M6 and M7 are involved in antagonist binding (β-receptor blockers). The cytoplasmic domains contain important binding sites for interactions with G proteins, as well as various kinases such as β-AR kinase (β-ARK)
15.3.3 Receptor Coupling
The G protein receptors interact intimately with G proteins. G proteins are complexes consisting of three subunits—α, β, and χ. There are different classes of G proteins, and attempts have been made to classify them as subtypes. For example, common α-subunits include: (1) αs (stimulatory) which activates adenylyl cyclase, (2) αi (inhibitory) which inhibits adenylyl cyclase, (3) α0 which modulates calcium channels and phospholipase C, and (4) αz which activates phospholipase C. In general, each α-subunit contains a region that interacts with the receptor, a site that binds guanosine triphosphate (GTP), and a site interacting with the effector system .
15.3.4 Receptor Function and Regulation
The traditional concepts of G protein-coupled receptor function are best illustrated in Fig. 15.1. First, an agonist binds to a receptor molecule, and then the seven-transmembrane-spanning receptor molecule interacts with a specific G protein; this, in turn, modulates a specific effector. Examples of typical agonists, receptors, G proteins, and effectors are provided in Fig. 15.1. While this illustration provides a general summary of the individual components needed for proper G protein-coupled receptor function, a more thorough understanding of the G protein receptor-mediated signaling, as well as its regulation, is best accomplished by the more specific example of the well-characterized beta-adrenergic receptor (β-AR) system. Therefore, we will start our discussion with these physiologically, pathophysiologically, and clinically highly relevant seven-transmembrane-spanning β-ARs .
15.4 Beta-Adrenergic Receptors (β-ARs)
15.4.1 Classification of β-Adrenergic Receptors
Table 15.2 summarizes the general features of β-AR subtypes identified to date; note that there are three subtypes [1]. While all of the subtypes can be found in the heart, the predominant subtype in the vasculature is the β2-AR. Importantly, norepinephrine and epinephrine are the endogenous agonists (catecholamines) that bind specifically to all three β-AR subtypes.
Table 15.2
Beta-adrenergic receptor subtypes : G proteins, tissue distributions, effectors, and signals
Receptor | β1 | β2 | β3 |
|---|---|---|---|
Primary G protein | Gs | Gs/Gi | Gs/Gi |
Tissue distribution | Heart | Vessels, heart, lung, kidney | Adipose, heart |
Primary effector in heart tissue | Adenylyl cyclase, L-type calcium channel | Adenylyl cyclase, L-type calcium channel | Adenylyl cyclase |
Signals | cAMP/PKA | cAMP/PKA, MAPK | cAMP/PKA |
Pharmacologically, β1- and β2-receptors are characterized by an equal affinity for the exogenous agonists isoproterenol and epinephrine, while norepinephrine (the neurotransmitter of the sympathetic nervous system) has a ten- to thirty-fold greater affinity for the β1-AR subtype. Also, β1-ARs are more closely located to synaptic nerve termini than β2-ARs, thereby being exposed and activated by higher concentrations of released norepinephrine .
15.4.2 β-AR Activation and Cardiovascular Function
Cardiac β-ARs are likely one of the most important types of receptor systems. β-ARs regulate important cardiovascular functions and are integral to the body’s flight or fight response. For example, during exercise, heart rate and contractility increase, cardiac conduction accelerates, and cardiac relaxation, an active process requiring adenosine triphosphate (ATP), is enhanced. Moreover, vascular relaxation (vasodilation) of many vascular beds can be observed during exercise (e.g., in skeletal muscles). All these effects are at least partially a direct consequence of β-AR activation and involve key elements of G protein receptor signaling: (1) receptor binding, (2) G protein activation, and (3) activation of an effector system. Importantly, most of these specific physiological cardiac effects are mediated via activation of β1-AR, while the vascular effects are mediated through the β2-receptor subtype. However, heart muscle also contains β2-receptors (as well as β3 receptors). Nevertheless, the β1-receptor subtype is the predominant cardiac isoform in healthy humans. More specifically, while there is a substantial population of β2-receptors in the atria, only around 20–30 % of the total β-AR population is β2-ARs in the left ventricle [2]. Not only is the activation of β-ARs and their associated signaling transduction critical in understanding the physiology of the cardiovascular system, but it has been recognized over the past decades that β-AR activations also play key roles in cardiac disease processes such as heart failure [3]. For example, an apparent paradox exists, namely, that β-adrenergic blockers (β-receptor antagonists) are beneficial in patients with heart failure; this will be discussed in more detail later in this chapter.
15.4.2.1 Effects of β-Receptor Activation on the Heart
β-Receptor activation on the heart regulates cardiac function on a beat-to-beat basis. Specifically, β-AR stimulation causes: (1) increases in heart rate (positive chronotropic effect), (2) increases in contractility (positive inotropic effect), (3) enhancements in cardiac relaxation (positive lusitropic effect), and/or (4) increases in conduction velocities (positive dromotropic effect). The molecular mechanisms leading to each of these specific effects will be discussed briefly. For further details, the reader is referred to the textbook by Opie [4]. Other important effects on cardiac myocytes that should be noted include those due to enhanced metabolism; the most significant effects on dilation in the coronary vasculature are mediated via metabolic waste products (CO2, etc.). Also see Chap. 21 for a detailed discussion of energy metabolism and feedback mechanisms .
15.4.2.2 Positive Chronotropic Effect
When activated, β-ARs located on the cells that make up the sinoatrial node increase the firing rate of the sinoatrial node. While the mechanisms of these effects are not fully understood, they are known to involve activation of G proteins and formation of cyclic adenosine monophosphate (cAMP), called a second messenger. Cyclic AMP can then bind to ion channels that are, in part, responsible for the diastolic (phase 4) depolarization of sinoatrial nodal cells. These ion channels, referred to as hyperpolarization-activated cyclic nucleotide-gated channels, have several unique properties, as they: (1) activate during hyperpolarization (more negative membrane potentials), (2) are responsive to cAMP, (3) are conductive for both Na+ and K+ ions, and (4) can be blocked by cesium. Cyclic AMP binding to the intracellular domain of these ion channels shifts their activation curves to more positive potentials, which increases the rates of “spontaneous” depolarization of sinoatrial nodal cells and ultimately their firing rates. The chronotropic effects can also be mediated via norepinephrine released from sympathetic nerve endings in the sinoatrial node and/or from circulating catecholamines (epinephrine and norepinephrine). From an integrative physiology standpoint, an increasing heart rate is an adaptive response to an increased demand of oxygen and cardiac output in the periphery. Accelerating the heart rate will, in turn, increase cardiac output if stroke volume remains constant (cardiac output = heart rate × stroke volume) .
15.4.2.3 Positive Inotropic Effects
As mentioned before, cardiac output needs to increase during exercise. One way to accomplish this is by increasing the heart rate; another is to increase stroke volume, e.g., by increasing cardiac filling (or increasing the preload). This can be accomplished by increasing the venous tone of the great veins that will increase filling of the heart chambers. It should be noted that this is an effect that is also mediated by adrenergic receptors but, in this particular case, by α-ARs causing venoconstriction (secondary to increased calcium influx). Such an increased preload then induces increased filling (and stretching) of the myocardial chambers, and via the Frank-Starling mechanism, stroke volume becomes augmented. However, stroke volume can also be efficiently improved by increasing the contractile force of the heart muscle at a given constant muscle length (i.e., independent of fiber length); this is called a positive inotropic effect and is mediated by activation of β-ARs, both β1-AR and β2-AR. The detailed molecular mechanisms involve the following processes (Fig. 15.3):
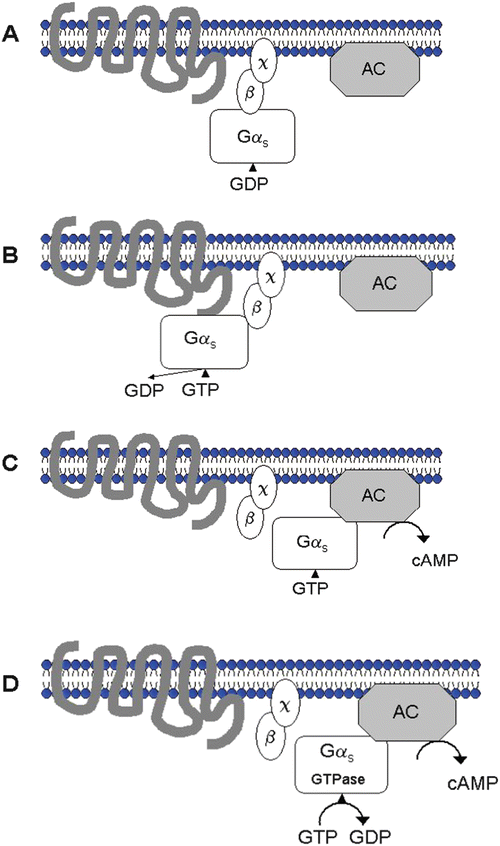
Fig. 15.3
G protein receptor activation and coupling with adenylyl cyclase (AC) during β-adrenergic receptor activation. Panel A: The receptor is inactive; GDP (guanosine diphosphate) is bound to α-subunit of stimulatory G protein. Panel B: The agonist binds to receptor which leads to receptor G protein interaction; GTP (guanosine triphosphate) binds to stimulatory G proteins (α-subunit, Gαs). Panel C: The G-subunits dissociate and Gαs stimulates adenylyl cyclase, with subsequent formation of cAMP (cyclic adenosine monophosphate). Panel D: Gαs GTPase becomes active, GDP reforms, and the activation cycle ends
1.
β-Agonist binds to either β1- or β2-receptor.
2.
GTP binds to the stimulatory α-subunit of the G protein (Gαs) and activates it.
3.
The G-subunits dissociate (the α-subunit dissociates from the β-subunit and χ-subunit).
4.
The α-subunit (containing GTP) stimulates an effector protein called adenylyl cyclase (AC).
5.
Cyclic 3′-5′ cAMP is formed and GTP is hydrolyzed to GDP (guanosine diphosphate) plus Pi (the α-subunit/GTP is a GTPase).
6.
The α-subunit/GDP binds to the β- and χ-subunits, and the initial (inactive) resting state reforms.
15.4.2.4 The Second Messenger Concept
cAMP is one of the key effector molecules in many of the signaling processes involving β-ARs (but also in many other biologic receptor systems). This molecule is commonly referred to as a second messenger and is one of many such molecules identified that are involved in cellular signal transduction. Another highly relevant second messenger is cyclic guanosine monophosphate (cGMP); cGMP is typically formed in vascular cells after binding nitric oxide, but can also be formed in cardiac and other cells after binding natriuretic peptides. As the name implies, these molecules trigger additional signaling mechanisms. Importantly, cAMP is present in all cardiac myocytes; it is rapidly turned over, and there is a constant dynamic balance between cAMP generation and breakdown via phosphodiesterases (enzymes breaking down cAMP). While cAMP can increase in response to β-adrenergic activation, there are other pharmacologically active agents that can lead to increased cAMP levels via alternate mechanisms. For example, glucagon is known to stimulate AC via a non-β-AR mechanism; forskolin stimulates AC directly; phosphodiesterase inhibitors such as milrinone, amrinone, and others inhibit the breakdown of cAMP resulting in increased cAMP levels. The exact mechanism of how formation of cAMP leads to increased cardiac contractility in cardiac myocytes will be further discussed below.
It is known that cAMP enhances the activity of protein kinase A. Once activated, protein kinase A causes the subsequent phosphorylation of numerous proteins including voltage-dependent sarcolemmal calcium channels, phospholamban (located in the sarcoplasmic reticulum), troponin I, and troponin C. The primary effect of phosphorylating the calcium channels and phospholamban is that this will increase calcium influx during a cardiac activation cycle and also increase calcium uptake by the sarcoplasmic reticulum. Working in concert, these effects will ultimately result in increased calcium transients during cardiac excitation-contraction coupling. Further, these increased intracellular calcium transients will result in increased ATP splitting by the myosin ATPase, which then increases the rate of development of contractile force (and also increases deinhibition of actin and myosin by the interaction of calcium with troponin C), ultimately causing an increase in total cardiac force development. If this stimulation is maintained, this effect or response often becomes attenuated over time. Also, cAMP levels may decrease, for example, due to activation of calmodulin that activates cAMP breakdown via phosphodiesterase activation and also via activation of β-AR kinase (β-ARK activation and downregulation).
It is important to recognize that the cellular and physiological responses to β-AR stimulation cannot simply be explained by overall increased tissue levels of cAMP. It has been speculated that cAMP may be compartmentalized in the heart, with a specific compartment available to increase contractility [5]. In addition, it has been shown that β2-ARs and β1-ARs, which both mediate positive inotropic responses, are so largely independent on cAMP generation. This latter response may, in some cases, be one of the myopathic mechanisms in chronic heart failure by resulting in increased systolic and diastolic calcium levels [6]. The complexity of the β-AR stimulatory G protein adenylyl cyclase signaling system can be further illustrated by the fact that there are about 20 identified Gα-subtypes, about 5 β-subtypes, and about 11 χ-subtypes, in addition to about nine isoforms of AC [7]. In cardiac tissue, isoforms V and VI of AC are considered to predominate. It should also be noted that specific anchoring proteins enable the juxtaposition of protein kinase A with its specific target proteins (A-kinase anchoring proteins), which may account for the resulting compartmentalization (and complexity) of responses .
15.4.2.5 Positive Lusitropic Effects
Phospholamban can be activated by protein kinase A (via cAMP) or by increased calcium levels subsequent to voltage-gated calcium channel activation (secondary to protein kinase A phosphorylation). This typical phosphorylation of phospholamban leads to increased activity of this enzyme, which then results in enhanced calcium removal from the cytosol back into the sarcoplasmic reticulum. It is known that the activation of troponin I, via protein kinase A, increases the rate of cross-bridge detachment and thus relaxation. Importantly, both of these responses will increase the rate of relaxation, an active process to remove calcium from the cytosol. A basic summary of the mechanisms of β-adrenergic increase in cardiac contractility and relaxation is provided in Fig. 15.4 .
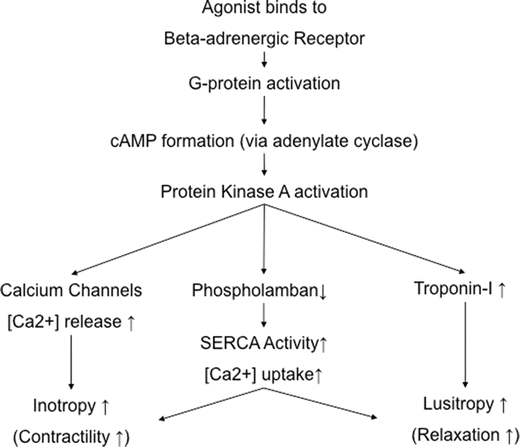
Fig. 15.4
The major intracellular signaling mechanisms involved in the regulation of cardiac contractility and relaxation following β-adrenergic receptor activation. It should be noted that there is also the possibility that L-type voltage-dependent calcium channels can be activated via a direct effect of a stimulatory G protein (Gαs) subsequent to β-adrenergic receptor (both β1- and β2-adrenergic receptors) activations. This overall signaling pathway is cAMP independent. The resulting increased systolic and diastolic calcium fluxes are considered to become a potentially myopathic mechanism in chronic heart failure
15.4.2.6 Dromotropic Effects
Activation of the voltage-gated (L-type) calcium channels in the atrioventricular node may enhance conduction in these nodal cells, as well as those of the Purkinje fibers .
15.4.2.7 Metabolic Effects
In general, the metabolic effects mediated via the β-AR system include (1) increases in glycogen formation (via increased glycogenolysis as well as decreased formation of glycogen), (2) the stimulation of lipolysis, and/or (3) increases in ATP production (via glycolysis/citrate cycle). Some of these metabolic effects may be mediated via the β3-AR (e.g., control of lipolysis). The β3-AR is also considered to have some regulatory function in cardiac (and vascular) contractility (e.g., negative inotropic effects) [8] .
15.4.2.8 Effects of β-Receptor Activation in the Vasculature
The main physiological function of β-AR activation in the vasculature is induced relaxation (vasodilation), which occurs via a reduction of cytosolic calcium levels in the smooth muscle cells (SMCs) composing these vessels, with relaxation having the greatest importance in arterioles. These relaxation responses are mediated via the β2-AR subtype and are antagonized by α-AR activation (α1-receptor subtype). The effects of α-receptor signal transduction will be discussed further below .
15.4.3 β-AR Regulation
To best understand β-AR regulation, it is useful to review the molecular structure of this receptor. Basically, the receptor is composed of three types of domains : (1) the extracellular domain, (2) the transmembrane domains which are involved in the agonist and antagonist binding (ligand binding), and (3) the intracellular or cytoplasmic domains which are important for G protein interactions and interactions with kinases such as β-ARK (β-adrenergic receptor kinase) (Fig. 15.2).
15.4.3.1 β-Adrenergic Receptor Desensitization and Downregulation
Receptor downregulation can be defined as a reduced presence in functional numbers of receptors in the cell membrane. Downregulation can result from internalization (and destruction in lysosomes), decreased rates of receptor degradation (nonlysosomal), and/or decreased synthesis. Desensitization is typically defined as receptor refractoriness, a “state of decreased activity following continued stimulation.” Yet, it should be noted that there is not always clear distinction between these two definitions in the literature. For example, the β-receptor can become desensitized during isoproterenol stimulation (a classic pharmacological β-adrenergic agonist). In contrast, during continuous β-AR stimulation, the enzyme β-ARK increases its activity; β-ARK phosphorylates the cytoplasmic domain sites of the β-AR (Fig. 15.2) and is able to uncouple the receptor from the Gαs (stimulatory G protein). This response acts as a block of the signal, and activation of adenylyl cyclase is decreased; in other words, adenylyl cyclase is uncoupled from the β-AR. Resensitization occurs once the receptor has been dephosphorylated; this mechanism is called agonist-specific desensitization. In contrast, non-agonist-specific desensitization can occur via second messenger cAMP (and also via diacylglycerol which activates protein kinases A and C) that phosphorylates the G protein-coupled receptors.
Specific examples of “physical” downregulation include internalization of receptors during continuous prolonged β-AR stimulation, which can be induced either pharmacologically (such as in an intensive care unit setting) or which is observed in heart failure (increased sympathetic tone and circulating catecholamines). This latter response may be a protective mechanism to minimize calcium overload of the cardiac myocytes secondary to β-adrenergic stimulation. Additional clinical examples of receptor regulation include “hypersensitivity” to β-AR agonists after propranolol (a β-AR blocker) withdrawal, following long-term use. This enhanced sensitivity is considered to be due to receptor externalization. Another example is drug tolerance to dobutamine (or other β-AR agonists); the molecular mechanisms underlying this clinical phenomenon may be desensitization.
While agonist-specific and unspecific desensitization can be viewed as one of the classical concepts of receptor regulation, G protein receptor kinases, such as β-ARK and associated proteins (β-arrestins), seem to function by other mechanisms. For example, β-arrestins are considered to interact with a G protein-coupled receptor and thus inhibit further G protein coupling after the receptor has been phosphorylated by β-ARK. While this more traditional role is now well established, β-arrestins have also been shown to be involved in receptor internalization as well as complex additional signaling mechanisms (e.g., mitogen-activated protein kinase pathways) [9, 10]. Internalization (also known as sequestration or endocytosis) is generally considered to serve various other functions including receptor resensitization (dephosphorylation) and recycling and/or altering signaling processes. Typically, the mechanisms of internalization involve the classical described clathrin-coated pit processes (caveolae) and also noncoated pit mechanisms. Another interesting role of β-arrestins is in ubiquitination; this is a means by which proteins are marked for subsequent degradation. For example, β-arrestin ubiquitination is important for β1-AR internalization, while receptor ubiquitination is necessary for lysosomal targeting and degradation of the receptors [11]. The gene regulation of the β-AR has been reviewed nicely elsewhere [7]) .
15.5 The Association Between β-ARs and Cardiac Disease
The function of the β-AR signaling pathway is not only of great importance in the normal regulation of physiological cardiac vascular function but has critical, although different, roles in the development of chronic heart failure. In general, heart failure is a syndrome defined by an inability of the heart to pump adequate amounts of blood to the body organs. The normal physiological response to an inadequate blood (oxygen) supply is to increase the cardiac output (and thereby oxygen delivery) via activation of the adrenergic nervous system (sympathetic nervous system); this response is characterized by an increase in circulating catecholamines. While this sympathetic response would intuitively be seen as beneficial, and indeed is also initially adaptive, in reality, the sustained β-AR activation and increased norepinephrine tissue levels [12] are considered to be associated with negative biological effects. Therefore, a sustained β-AR response in the cardiac patient may be, in fact, considered both maladaptive and inappropriate. Consistent with this notion, adrenergic drive is increased in all chronically failing human hearts with systolic dysfunction, as well as in all animal models of hemodynamic overload. Thus, adrenergic drive can also be considered as a servo-control mechanism to maintain cardiac performance at an acceptable level [7]. For example, the proportion of β1- to β2-adrenergic subreceptor population in normal human ventricles shifts from 70:30 in diseased human ventricles to about 50:50 in normal human ventricles [1]. Also on the molecular level, both receptor subtypes are typically desensitized, due to uncoupling of the receptors from their signaling pathways associated with both increased β-AR phosphorylation and upregulation of the inhibitory G protein, Gi.
To illustrate the maladaptive β-AR signaling response, it is important to consider that β-adrenergic agonists, given to patients with chronic heart failure, may actually increase mortality. In contrast, the administration of β-blockers has been shown to improve patient survival; β-AR blockers continue to be one of the cornerstones of medical/drug therapy for patients with chronic heart failure.
In certain patients with cardiac failure, there is a situation where increased adrenergic drive is required to maintain the circulatory needs of their bodies; the more the heart is activated adrenergically, the more profound are the effects of desensitization which, in turn, activates the adrenergic system even more, ultimately causing myocardial tissue damage. Based on the studies in which β-AR blocking agents were administered, it is generally believed that the β-AR system is central in this vicious pathological cycle (or positive feedback loop), which is commonly characterized by progressive remodeling and myocardial dysfunction.
15.5.1 Beta-Adrenergic Signaling and Heart Failure
One of the hallmarks of chronic heart failure is adrenergic overdrive leading to β-AR downregulation and desensitization. In particular, β1-ARs have been shown to be downregulated selectively, while the β2-ARs are relatively increased so that, as mentioned above, the population of β1 to β2 shifts from 75/25 to about 50/50. The marked uncoupling of the β-ARs from the G proteins is due to increased levels of β-ARK1, as well as an increased inhibitory G protein (Gi) [14]. This, in turn, results in not only attenuated β-AR signaling but also activation of additional pathways involved in ventricular remodeling.
The shift of the β1-/β2-AR subpopulation to an increased number of β2-receptors is only one of the phenomena associated with chronic heart failure. More specifically, β2-ARs couple to a number of signaling pathways, including a potential coupling to an inhibitory G protein (Gαi). Also, β2-ARs may modulate several G protein-independent pathways, including inhibition of sodium-hydrogen exchanges as well as a nonprotein kinase A-dependent interaction with L-type calcium channels [6, 15]. There are other G protein-independent pathways in which the β2-ARs have been implicated, such as those associated with phosphatidylinositol kinase, phospholipase C/protein kinase C, and/or arachidonic acid signaling. And while many details of these pathways remain poorly understood, it is becoming more clear that some of these pathways play important roles in the molecular pathobiology of heart failure.
One of the widely recognized mechanisms of β-AR desensitization is agonist-dependent β-ARK-mediated phosphorylation of the β-AR itself. Moreover, the beta-gamma subunits of the G proteins can act as signaling molecules themselves, with the potential of activating many downstream targets of pathways such as adenylyl cyclases, PLC, PLA2, PI3K, K+ channels, and/or Src-Ras-Raf-MEK-MAPK (mitogen-activated protein kinase) [7].
The potentially toxic effects of long-term β1-AR activation are also illustrated by the fact that selective β1-AR agonist or receptor overexpression leads to increased cardiac apoptosis or programmed cell death [16–18]. The mechanisms by which such apoptosis is activated have been attributed, in part, to initial activation of calcineurin via increased intracellular calcium through L-type calcium channels [19]. Additionally, direct toxic effects of catecholamines on the myocytes have been described as a result of direct β-AR activation [12]. Other effects of chronic β-AR signaling include production of cytotoxicity via calcium overload, increased free radical formation, as well as stimulation of pathologic hypertrophy [7]. For example, the marked uncoupling of β1- and β2-receptors from their physiological signaling pathways not only results in attenuation of the β-AR signaling but also allows for the coupling of the receptors with other possible myopathic signaling pathways involved in ventricular remodeling (e.g., MAPK and PI(3)K cascades). As stated earlier, β-receptor antagonists may be a beneficial therapy in the chronic heart failure patient. Specifically, the β1-specific β-AR blocking drug metoprolol produces reverse remodeling similar to a combined α-β-blocker carvedilol, suggesting that β1-AR signaling is an important determinant of pathologic hypertrophy in human heart failure [20].
Lastly, genetic heterogeneity in expression of the β-AR in the general population has been identified which may be associated with disease susceptibility. For example, a substitution of threonine to isoleucine at amino acid 164 in the β2-AR has been linked with reduced survival and exercise tolerance in patients with heart failure [21, 22].
A summary of the known changes in β-AR signaling is provided in Table 15.3. Lastly, it should be mentioned that investigations using transgenic animal models have greatly helped to elucidate some of the important disease mechanisms associated with adrenergic receptor reception pathways; they have also served as tools in identifying and testing novel or evolving therapeutic targets for the treatment of heart failure [1] .
Table 15.3
β-Adrenergic receptor signaling pathways in heart failure
Molecule | Change |
|---|---|
β1-AR | ↓, uncoupled |
β2-AR | NC, uncoupled |
β-ARK1 | ↑ |
β-Arrestins 1 and 2 | NC |
Galpha1 | ↑ |
15.6 Alpha-Adrenergic
15.6.1 Physiology
Two major α-AR subtypes have been identified—α1 and α2. The α1-type receptors have been described to be present in the heart, as well as in the vessels and smooth muscle. The primary response to their activation in cardiac cells is phospholipase C-β which leads to an increase in diacylglycerol and inositol triphosphate (InsP3) and also activates both protein kinase C and MAPKs. Physiological receptor agonists include norepinephrine and epinephrine, and a Gq protein is the primary associated G protein [23]. In general, the primary physiological importance of α1-receptors is far greater in the vasculature, leading to calcium influx and vasoconstriction via the second messenger InsP3. In cardiac muscle cells, α1-receptors have been shown to induce a positive inotropic effect; like β-receptors, this response is also associated with formation of InsP3 [24]. Alpha-2-receptors have been identified on coronary vessels, as well as in the central nervous system (presynaptic); they reduce the activity of AC (and protein kinase A) via an inhibitory G protein.
In summary, the primary physiological role of α1-receptors is for the vasculature regulation of tone via vasoconstriction, which is secondary to calcium influx into the SMCs of the circulatory arterioles.
15.6.2 Role in Disease States
In general, in heart failure patients, the α1-AR system will elicit an increase in receptor density and is also characterized by changes in inotropic responses [25]. In addition, α1-AR activation has been shown to promote cardiomyocyte hypertrophy via activation of hypertrophic MAPK pathways) .
15.7 Role of Adrenergic and Other Receptors in Myocardial Hypertrophy
Prolonged or repeated increased workloads on the heart lead to cardiac hypertrophy. Hypertrophy is typically characterized by increased contractile protein content (and muscle mass), myofilament reorganization, and reexpression of embryonic markers. As discussed earlier, myocyte cell growth by G protein-coupled receptors may involve not only Ras and MAPK pathways but also other signaling pathways (e.g., calcineurin, PI3K, and phosphatidylinositol-3-OH). Abnormal β-AR function with elicited hypertrophy is also characteristic; in particular, elevated myocardial levels of β-ARK1 can be detected [26]. As mentioned above, α1-receptor signaling also may be very important in the development of cardiac hypertrophy. Specifically, Gq-coupled receptors, such as angiotensin, α1-AR, and endothelin, can all activate hypertrophic MAPK pathways [27, 28]. To summarize, the cardiac hypertrophic response may not be compensatory to reduce wall stress but rather maladaptive and thus increase mortality. In particular, signaling pathways involving elevated catecholamine levels (norepinephrine and epinephrine) and activated Gq-coupled receptors seem to promote hypertrophic responses. Thus, targeting these specific signaling pathways may be a novel therapeutic approach for the potential treatment of “maladaptive” cardiac hypertrophy and ultimately heart failure. For further details on this topic, the reader is referred to the review by Rockman et al. [1] .
15.8 Muscarinic Receptors
15.8.1 Physiology
The muscarinic receptor system in the heart is also an associated G protein-coupled receptor system, one fairly similar to the β-AR. The main cardiac receptor subtype (M2 receptor) is a cholinergic receptor and thus mediates primary parasympathetic (syn. nervus vagus, cholinergic, acetylcholine mediated) nervous system responses. For more details, refer to Chap. 14. Its general function is to simultaneously balance and antagonize the sympathetic effects of β-AR activation, with its most pronounced effects on the controls of the sinoatrial (heart rate, chronotropic effect) and atrioventricular nodes (conduction, dromotropic effect). There exist very few muscarinic M2 receptors in the ventricles, so activated negative inotropic (contractility) effects subsequent to M2 receptor activation are very small. The primary control signal mediated via the vagus nerve is that which leads to a local release of acetylcholine in the sinoatrial and atrioventricular nodes. Acetylcholine then binds to the M2 receptor, activates an inhibitory G protein (Gαi), and essentially decreases the activity of adenylyl cyclase, which directly leads to opening of K+ channels. Thus, in the sinoatrial node, vagal stimulation tends to a flattening of the diastolic depolarization, which then induces a slowing of heart rate (bradycardia, negative chronotropic effect) possibly via the effects of reduced cAMP availability on If current (hyperpolarization-activated cyclic nucleotide-gated channel) but also via activation of a potassium outward current which in concert decreases the spontaneous firing rate of the sinoatrial node. In the atrioventricular nodal tissue, vagal stimulation also activates an inhibitory G protein, which typically causes a slowing conduction velocity via a decreased calcium influx through L-type calcium channels. Clinically, the effects of vagal stimulation on the atrioventricular node are detected as increased atrioventricular nodal conduction times (e.g., prolonged PR interval).
The M3 muscarinic subtype is primarily found in vascular SMCs. Its activation leads to contraction of vascular smooth muscles via a Gq/11-PLC-InsP3-mediated calcium influx (as well as diacylglycerol/phospholipase C/protein kinase, cAMP elevation) .
15.9 Other G Protein-Coupled Cardiovascular Receptors
There are other G protein-coupled cardiovascular receptor systems which have important roles in cardiac physiology and pathophysiology, yet it is beyond the scope of this chapter to review all these systems in detail. However, for some degree of comprehensiveness, a current list of important G protein-coupled receptor systems and their respective functions is provided in Table 15.4 .
Table 15.4
Other cardiovascular G protein-coupled receptor systems
Family | Type | Cardiovascular function | G protein | Signaling |
|---|---|---|---|---|
Adenosine | A1 | Bradycardia | Gi/o | AC inhibition; K+ opening |
A2A | Vasodilation | Gs/G15 | AC, PLCβ | |
A2B | Vascular smooth muscle relaxation | Gq/11? | PLC/AC-mediated calcium activation | |
A3 | A1 modulation | Gi3 | AC inhibition; PLC, InsP3-mediated calcium increase | |
Angiotensin | AT1 | Vascular smooth muscle contraction; cell proliferation; cell hypertrophy; antinatriuresis | Gq/11 | PLC/PKC-mediated calcium elevation |
AT2 | Vasodilation; apoptosis; growth inhibition; natriuresis; nitric oxide production | Gia2/Gia3 | MAPK activation; K+ opening; PTPase activation leading to T-type calcium channel closure | |
Endothelin | ETA | Vasoconstriction | Gq/11 | PLC/InsP3; cAMP |
ETB | Vasodilation/vasoconstriction (ETB2) | Gi; Gq/11 | cAMP inhibition; PLC/InsP3 calcium increase | |
Opioid | OP1 (delta), OP3 (mu) | Central cardiovascular regulation (OP1, 3); cardioprotection (OP1) | Gialpha1/3; Go | IK conductance activation; reduction in neuronal Ica; AC inhibition; PKC and KATP channel activation (cardioprotection) |
15.10 Receptor Cross Talk
Cross talk between receptors refers to a biological phenomenon whereby interactive regulatory (or modulatory) messages are sent from one receptor to another. Many of the aforementioned receptor pathways are engaged in some degree of cross talk, which is commonly via second messengers and/or protein kinases which, in turn, modulate G protein-mediated messages. While the current understanding of receptor cross talk is far from being complete, it is clear that receptor cross talk is integral for sustaining a coherent function of the cardiovascular system and has implications in both normal physiology and elicited cardiac disease states.
The following is an example of one such cardiovascular cross-talk interaction and its relevance to the pharmacological treatment of disease. Heart failure is generally characterized by dysfunction of the β-AR system (downregulation of β1-receptors, uncoupling of β-AR and AC) but also the renin-angiotensin system (downregulation of AT1 receptors); distinct signaling pathways have been identified which then characterizes both β-AR (e.g., via Gs and AC) and AT1 receptor signaling (e.g., via Gq/11 and PLCβ). In a recent study, it was shown that selective blockade of β-AR inhibited angiotensin-induced contractility, while selective blockade of AT1 receptors reduced catecholamine-induced reduction in heart rate [29]. These unusual transinhibitory effects were considered to be caused by underlying G protein receptor uncoupling. Furthermore, direct interactions of these receptor systems were demonstrated in vivo, and it is speculated that such interactions may have a profound role in determining the ultimate responses to drugs designed to block a given receptor subtype (e.g., β-AR blockers, AT1-receptor blockers). Note that cardiovascular receptor cross talk has been extensively reviewed by Dzimiri [30].
15.11 Guanylyl Cyclase-Linked Receptors
15.11.1 Soluble Guanylyl Cyclase (Receptor for Nitric Oxide)
Although structurally different, the general principles of receptor signaling as discussed for the β-AR apply also for nitric oxide/guanylyl cyclase signaling. Specifically, the agonist nitric oxide (NO) is considered as a universal signaling molecule present in all tissues, and the role of NO signaling is similar to other signal transduction systems, not only important in normal cardiac function but also in disease processes. Physiologically, nitric oxide is produced by several isoforms of the enzyme nitric oxide synthase (NOS). To date, three isoforms have been described to function within the cardiovascular system—eNOS, iNOS, and nNOS. Nitric oxide is a small, lipid-soluble, gas molecule that readily crosses cell membranes. Inside the cell, it typically binds to a receptor, the soluble (cytoplasmic) guanylyl cyclase (GC, an enzyme which converts GTP to cyclic 3′-5′ GMP). Cyclic GMP is the second messenger in this signaling cascade and, in the cardiovascular system, can activate two major effector systems—cGMP-regulated phosphodiesterases and cGMP-dependent protein kinases (cGKs).
In the vascular SMCs, NO inhibits vascular smooth muscle constriction (causes vasodilation) via inhibition of cytoplasmic calcium release. However, the underlying mechanisms for the important vasodilatory effect of NO are complicated and minimally involve a cGMP-dependent protein kinase (cGKI); these pathways have been reviewed in detail elsewhere [31]. Nitric oxide has also been shown to reduce vascular smooth muscle proliferation (clinically important in in-stent restenosis) and migration. It is well established that NO release may reduce platelet adhesion and activation, as well as vascular inflammation [32]. Importantly, NO insufficiencies are considered to be an important factor in endothelial dysfunction, leading to the increased susceptibility for arterial thrombosis formations [33].
Nevertheless, the relevance of NO signaling is not limited to vascular biology or pathobiology; it has been demonstrated that NO has an important pathophysiological role in heart failure as well, i.e., in the modulation of cardiac function, cardiovascular protection, and/or in the regulation of apoptosis [34–36]) .
15.11.2 Membrane Guanylyl Cyclase-A (Receptors for Natriuretic Peptides)
To date, at least seven membrane-bound enzymes synthesizing cyclic GMP (cGMP) have been identified. All seven of these membrane guanylyl cyclases (GCs) have a common structure (Fig. 15.5). For the purpose of this discussion, guanylyl cyclase-A has the greatest importance relative to the heart and will be discussed in detail .
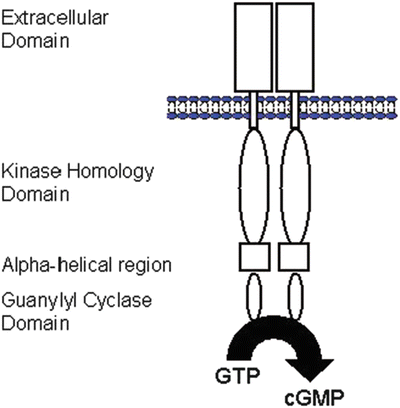
Fig. 15.5
Basic topology of membrane GC-A receptor. The membrane GC (guanylyl cyclase) form homodimers or higher-order structures (modified from [37]). cGMP cyclic guanosine monophosphate, GTP guanosine triphosphate
15.11.3 Physiology
Currently, three natriuretic peptides have been identified—ANP, BNP, and CNP. Both ANP and BNP bind to GC-A and are released in the heart; they are considered as cardiac hormones. In general, CNP is mainly produced by the vascular endothelium and may be a regulator of vascular tone and cell growth through GC-B activation. This receptor consists of an extracellular domain, a kinase homology domain, an α-helical amphipathic region (hinge region), and a C-terminal guanylyl cyclase (catalytic) domain (Fig. 15.5). This receptor is considered to exist in dimers (two molecules coupled together).
The major effects of GC-A are via the formation of, and the activation by, cGMP. Since, in general, the agonists are circulating hormones (peptides), a variety of organ systems can be affected simultaneously following ANP/BNP release. ANP is mainly produced in the atrium, while BNP is produced primarily in the ventricles. The primary triggering factors for ANP/BNP release are wall stretch and/or pressure increases, but their release may also involve neurohumoral factors (glucocorticoids, catecholamines, angiotensin II, etc.). The primary effect of ANP/BNP release is modulation of blood pressure/volume; the half-lives of ANP/BNP are around 2–5 min. Interestingly, although both ANP and BNP bind to the same receptors, it has been shown that targeted genetic disruption of ANP or BNP in mice induces a unique phenotype; ANP-deficient mice show arterial hypertension, hypertrophy, and cardiac death. In contrast, the BNP-deficient mice phenotype is characterized by mostly cardiac fibrosis. More specifically, BNP has a lower affinity to GC-A than ANP and may act mostly as a local paracrine (antifibrotic) factor. Also, under physiological conditions, the ANP levels are much higher than BNP levels in the circulating blood, and the potency for induced vasorelaxation is also less for BNP vs. ANP [37].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


