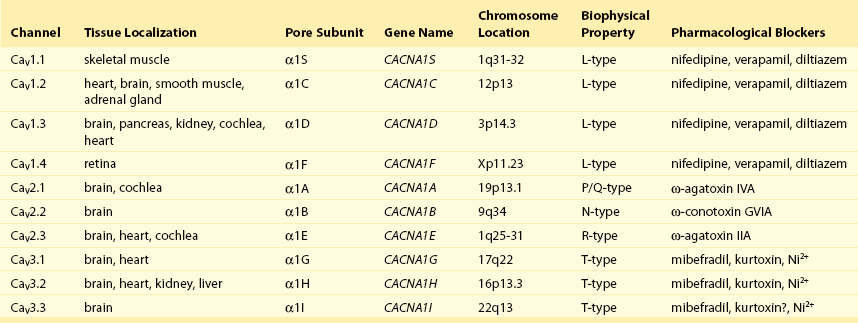
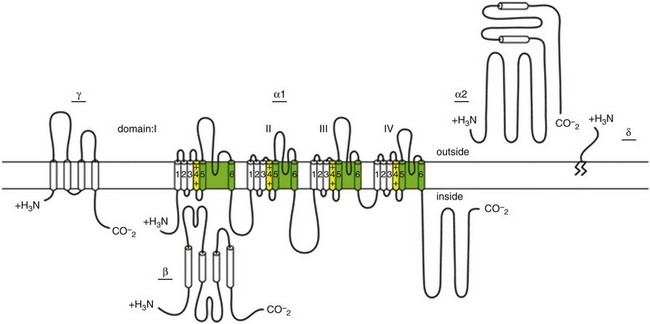
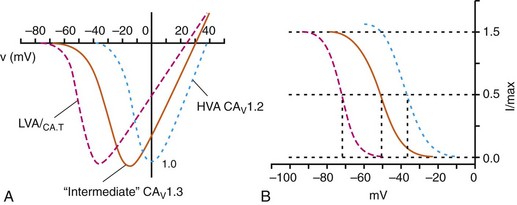
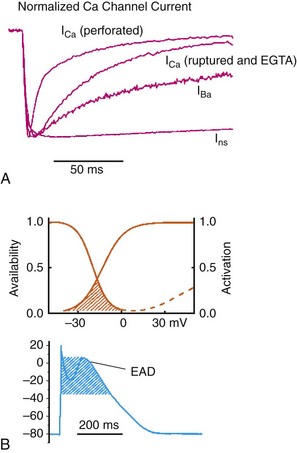
2 Calcium (Ca2+) permeable ion channels are involved in a number of fundamental processes in the heart, including automaticity in the sinoatrial (SAN) and atrioventricular (AVN) nodes, excitation-contraction coupling in the working myocardium, and regulation of gene expression in hypertrophic signaling.1–4 Several ion channels in the plasma membrane of cardiac myocytes and cardiac fibroblasts are involved in transporting Ca2+ into the cytosol from the extracellular space (Table 2-1). Included among these are two forms of L-type Ca2+ channels (LTCC; CaV1.2 and CaV1.3), T-type Ca2+ channels, and several transient receptor potential (TRP) channels. These different Ca2+ permeable ion channels play distinct roles in different parts of the heart. For example, CaV1.2-mediated L-type Ca2+ current (ICa,L) is present in all cardiomyocytes, whereas CaV1.3-mediated ICa,L is restricted to SAN, AVN, and working atrial myocardium. T-type Ca2+ currents (ICa,T) are mainly expressed in the SAN and AVN in normal conditions, but may be expressed in ventricular myocytes in cardiac hypertrophy. In most cases, TRP channels conduct nonselective cation currents that include Ca2+ influx, which may be important in both cardiac myocytes and cardiac fibroblasts, the latter of which don’t typically express robust voltage-gated Ca2+ channels. The purpose of this chapter is to review the expression patterns, biophysical properties, and structure-function relationships of Ca2+ permeable channels in the heart. Table 2-1 Nomenclature of the Voltage-Gated Ca2+ Channels Along With Chromosome Location and Biophysical and Pharmacological Properties LTCCs are multimeric proteins consisting of an α1-subunit that constitutes the pore of the channel and several accessory subunits denoted β, α2-δ, and γ2,5,6 (Figure 2-1). Currently, four α1-subunits for L-type Ca2+ channels are known, and two of these, CaV1.2 (α1C encoded by the CACNA1C gene) and CaV1.3 (α1D encoded by the CACNA1D gene), are expressed in the heart3,5,7 (see Table 2-1). These α1-subunits form the channel pore and contain the voltage sensor that controls channel gating, as well as drug binding sites and regulatory sites targeted by second messengers. Ca2+ channel α1-subunits have a similar structure to voltage-gated Na+ and K+ channels, whereby they are organized into four domains (I through IV), each containing six transmembrane segments (S1 through S6). The voltage sensor is located in the S4 transmembrane segment of each domain, and the pore loops are located between S5 and S6. Alternative splice variants of CaV1.2 have been documented and can impact regulation by second messengers and drug binding. Recently, alternative splice variants of CaV1.3 have also been described.8,9 These variants result in the expression of long and short forms of CaV1.3, which are characterized by differences in biophysical properties such as the activation midpoint; however, not all of the short forms of CaV1.3 are expressed in the heart.8 Figure 2-1 Ca2+ channel subunit structure. L-type Ca2+ channels consist of the pore-forming α1-subunit and a series of accessory subunits (β, γ, α2δ). Predicted α-helices are depicted as cylinders. The lengths of the lines correlate with the approximate length of the polypeptide segments. T-type Ca2+ channels consist of the pore-forming α1-subunit; however, whether accessory subunits associate with T-type Ca2+ channels in native cardiomyocytes is less clear. (From Catterall W: Voltage-gated calcium channels. Cold Spring Harb Perspect Biol 3:a003947, 2011.) Ca2+ channel β-subunits (CaVβ1 through CaVβ4) are encoded by four genes (CACNB1-4) and, like the α1-subunits, can be alternatively spliced to generate a number of isoforms.10,11 Crystal structures of LTCC β-subunits demonstrate that these subunits contain conserved Src homology 3 (SH3) and guanylate kinase domains, as seen in scaffolding proteins in the membrane-associated guanylate kinase (MAGUK) family.12,13 CaVβ-subunits are intracellular proteins (see Figure 2-1) that bind to a single site, called the α interaction domain (AID), on the loop connecting domains I and II on the α1-subunit of the LTCC. CaVβ acts as a chaperone protein that facilitates the trafficking of LTCCs to the plasma membrane and also modulates the biophysical properties of these channels. Specifically, coexpression of CaVβ2 with CaV1.2 increases ICa,L amplitude, accelerates activation and inactivation kinetics, and shifts the steady inactivation curve to more negative membrane potentials. CaVβ also profoundly enhances the affinity of dihydropyridines (blockers of LTCCs) for the α1-subunit by decreasing their dissociation from the channel.14 Four genes (CACNA2D1-4), which can be alternatively spliced, encode the α2-δ-subunit of the LTCC.15,16 The α2-δ1 and α2-δ3 isoforms are expressed in the heart. The protein is cleaved post-translationally and then is relinked via disulfide interactions. This subunit is primarily extracellular, with the δ-subunit portion anchored in the plasma membrane (see Figure 2-1). CaVα2-δ-subunits facilitate the targeting of LTCCs to the plasma membrane and increase ICa,L by shifting the voltage dependence of activation and inactivation to hyperpolarized membrane potentials. Ca2+ channel γ-subunits are membrane-bound proteins encoded by eight genes (CACNG1-8) with several isoforms, including γ4, γ6, γ7, and γ8, which are present in cardiac muscle. These isoforms associate with CaV1.2 and alter activation and inactivation properties of the channel.17 As their name suggests, LTCCs are highly selective for Ca2+ over monovalent cations. The channel pore is approximately 6 Å in diameter at its narrowest point,18 indicating that size is not the main factor in determining selectivity. Rather, a series of four glutamate residues (EEEE motif) is responsible for conferring ion selectivity through the ability of this motif to bind divalent cations (Ca2+ and Mg2+), which block the passage of monovalent cations.19,20 As additional divalent ions enter the channel, bound Ca2+ ions are displaced from the EEEE motif and are pushed through the pore, which generates ICa,L. At the single-channel level, ICa,L conductance in ventricular myocytes (i.e., CaV1.2 dependent) has been reported to be approximately 5 pS in 2 mM Ca2+ and approximately 15 pS with Ba2+ as the charge carrier.21 Similarly, single-channel conductance for CaV1.3 has been determined in heterologous expression systems and has been found to be approximately 15 pS with Ba2+ as the charge carrier.8 ICa,L is mediated by CaV1.2 and CaV1.3 in the heart, and these two channel isoforms are distinguished by their unique biophysical properties7,22 (Figure 2-2). CaV1.2-mediated ICa,L, which is expressed throughout the myocardium (atrium, ventricles, and conduction system), has a bell-shaped current-voltage (I-V) relationship in which the current activates at membrane potentials positive to −40 mV and peaks between 0 and +10 mV. The V1/2 of channel activation (V1/2(act) ) is typically between −10 and −15 mV. In contrast, recombinant CaV1.3–dependent ICa,L activates at more negative membrane potentials (i.e., the V1/2(act) is hyperpolarized) and displays slower inactivation kinetics.8,23,24 CaV1.3 is also less sensitive to dihydropyridines than is CaV1.2. Figure 2-2 Voltage dependence of activation and steady state inactivation of ICa,T, CaV1.3-mediated ICa,L, and CaV1.2-mediated ICa,L from mouse SAN. A, Current-voltage (I-V) relationships illustrating that ICa,T activates most negatively, and CaV1.2-mediated ICa,L activates most positively. CaV1.3-mediated ICa,L has intermediate activation properties. B, Steady state inactivation curves for ICa,T, CaV1.3-mediated ICa,L, and CaV1.2-mediated ICa,L. Dashed lines indicate the voltage at which 50% of channels are inactivated (V1/2(inact) ) for each Ca2+ channel. Dotted lines indicate points of complete current inactivation and full channel availability. (From Mangoni MM, Couette B, Marger L, et al: Voltage-dependent calcium channels and cardiac pacemaker activity: from ionic currents to genes. Prog Biophys Mol Biol 90:38, 2006.) CaV1.2 is the primary determinant of ICa,L in the ventricular myocardium, where it plays a prominent role in excitation-contraction coupling and the Ca2+ induced–Ca2+ release process.1 Approximately 75% of these LTCCs are located in dyads, in close proximity to the ryanodine receptors located in the junctional sarcoplasmic reticulum, within the t-tubular system (see Figure 2-4). Consistent with the critical role of CaV1.2 in the ventricles, CaV1.2 knockout mice die before birth in association with cardiac failure.25 CaV1.2 is also expressed in the SAN, AVN, and atrial myocardium, along with CaV1.3.22,26 As a result, total ICa,L in these supraventricular tissues is dependent on both α1C and α1D pore-forming subunits and demonstrates biophysical characteristics that are different from those of the ventricles. The generation of CaV1.3 knockout mice has been important in determining the biophysical properties of these channels and in distinguishing them from CaV1.2-mediated ICa,L (and ICa,T).23,27 Specifically, these CaV1.3 knockouts have been used to show that ICa,L in SAN and atrial myocytes activates between −60 and −50 mV in association with a left shift in the V1/2(act) and peaks at membrane potentials around −10 mV. The steady state ICa,L inactivation curve is also shifted to the left when CaV1.3 contributes to total ICa,L. Together, available data show that CaV1.3-dependent ICa,L has intermediate biophysical properties compared with CaV1.2-dependent ICa,L and ICa,T. Figure 2-4 Subcellular localization of L-type Ca2+ channels in cardiomyocytes. LTCCs associate with anchoring proteins (AKAPs) and other signaling proteins to create distinct populations of LTCCs in T-tubules and within caveolae. Also, clustering of LTCCs in discrete regions of the plasma membrane results in coupled gating. See text for details. (From Best JM, Kamp TJ: Different subcellular populations of L-type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol 52:376-387, 2012.) The unique biophysical properties of CaV1.3 enable ICa,L to play a prominent role in pacemaker activity in the SAN and in the electrical conduction in the atrial myocardium. In the SAN, CaV1.3-mediated ICa,L contributes to the diastolic depolarization phase of the action potential and thus is a major determinant of heart rate in vivo. CaV1.2, on the other hand, contributes more prominently to the action potential upstroke in SAN myocytes. Consistent with this, CaV1.3 knockout mice display sinus bradycardia in association with a reduced diastolic depolarization slope.26–28 CaV1.3 knockout mice also show increased susceptibility to atrial fibrillation,24,29 a very common cardiac arrhythmia, confirming that these Ca2+ channels play an important role in atrial conduction. ICa,L undergoes decay during sustained depolarization, which is termed inactivation (Figure 2-3, A). This inactivation process is time, voltage, and calcium dependent.30–32 Voltage-dependent activation is relatively slow, as is indicated by the degree of inactivation with Ba2+ as the charge carrier, or when monovalent cations permeate LTCCs in the absence of divalent cations.33 In the presence of extracellular Ca2+, inactivation is much faster, and this Ca2+-dependent inactivation is further accelerated in the presence of functional SR Ca2+ release (i.e., during normal excitation-contraction coupling).34 This indicates that both the Ca2+ entering the myocytes through the LTCC and the Ca2+ released from the SR during the Ca2+ transient contribute to LTCC inactivation. Figure 2-3 A, Ca2+ channel inactivation with Ca2+, Ba2+, or monovalent cations (ns) as the charge carrier. Currents were measured at 0 mV except for Ins at −30 mV to obtain comparable activation, and peak currents were normalized. ICa with SR Ca2+ release (i.e., Ca2+ transients occur) was recorded using the perforated patch-clamp technique and 2 mM external Ca2+. ICa with no SR Ca2+ release (i.e., no Ca2+ transients occur) was recorded in the whole-cell configuration with 10 mM EGTA in the pipette. IBa was recorded in the whole-cell configuration with 2 mM external Ba2+ and 10 mM EGTA in the pipette. Ins was measured in divalent cation-free conditions. The t1/2 of current decline progressively increases from top to bottom, illustrating that ICa inactivation is both Ca2+ and voltage dependent. B, Overlap of steady state activation and inactivation curves for ICa,L illustrates the presence of a window current (dashed lines), which may contribute to the generation of early afterdepolarizations (EADs). ICa,T can also produce window currents at more negative membrane potentials (between −80 and −40 mV). (A, From Bers DM, Perez-Reyes E: Ca channels in cardiac myocytes: structure and function in Ca influx and intracellular Ca release. Cardiovasc Res 42:339-360, 1999. B, From Benitah JP, Alvarez JL, Gomez AM: L-type Ca2+ current in ventricular cardiomyocytes. J Mol Cell Cardiol 48:26-36, 2010.) Ca2+-dependent inactivation of ICa,L is a calmodulin (CaM) dependent process.35,36 CaM is bound to the C-terminus of the channel via the IQ domain, and when local Ca2+ is elevated, an increase in Ca2+ binding to CaM occurs. This enhances the interaction of CaM with the IQ domain, thereby causing inactivation of the channel. Ca2+-dependent inactivation may serve as a protective negative feedback mechanism to prevent Ca2+ overload in cardiomyocytes. Steady state inactivation (i.e., channel availability), like voltage-dependent activation, follows a sigmoidal relationship, with a V1/2(inact) of ≈−35 mV for CaV1.2-dependent ICa,L and −45 mV for CaV1.3-dependent ICa,L.22 The activation and inactivation curves for ICa,L can overlap, resulting in a “window current,” which has been postulated to play a role in the generation of arrhythmogenic early afterdepolarizations (EADs; see Figure 2-3, B).37,38 LTCCs are also known to undergo a process called facilitation, whereby a progressive increase in ICa,L amplitude and the time constant of inactivation can occur during increases in pacing frequency.34,39 The facilitation process occurs as the result of a reduction in Ca2+-dependent inactivation and is mediated by calmodulin-dependent kinase II (CaMKII) phosphorylation.40 It is now appreciated that LTCC can be organized into distinct subcellular compartments through the actions of scaffolding proteins, including A-kinase anchoring proteins (AKAPs),41,42 within cardiomyocytes43 (Figure 2-4). Examples of subpopulations of LTCCs include those found in dyads within the T-tubules and those in separate plasma membrane domains such as caveolae and lipid rafts.2 This pattern of organization is critical for the precise spatio-temporal modulation and regulation of LTCCs in different physiological functions such as excitation-contraction coupling, transcriptional regulation, and responses to β-adrenergic (β-AR) receptor activation. Within dyads, LTCCs from a complex with β-ARs, adenylyl cyclase (AC), protein kinase A (PKA), and AKAPs.2,44,45 These proteins area also in close apposition to a complex of proteins associated with the SR, including ryanodine receptors, PKA, protein phosphatase 2A (PP2A), phosphodiesterases, and AKAPs (AKAP5 and AKAP15).41,42 It is the AKAPs that are thought to be responsible for the organization of these signaling complexes. Within caveolae, LTCCs associate with β2-ARs, AC, PKA, PP2A, and caveolin 3, all of which enable LTCCs in caveolae to be locally stimulated by β2-AR activation.46 LTCC also demonstrate a property called coupled gating, which occurs when LTCCs are grouped together into clusters in the plasma membrane, resulting in localized regions with significantly elevated intracellular Ca2+ concentrations. Coupled gating is enhanced by the scaffolding protein AKAP5 and by protein kinase C (PKC), which form the signaling complex with the clusters of LTCCs.43,47 Although L-type Ca2+ channels get the lion’s share of the attention in both cardiac and vascular electrophysiology, low voltage-activated T-type Ca2+ channels also play important roles in normal and diseased hearts. Three T-type Ca2+ channels exist in mammals: Cav3.1 (α1G), Cav3.2 (α1H), and Cav3.3 (α1I), with Cav3.1 and Cav3.2 channels being the major channels in heart48,49 (see Table 2-1). Although L-type Ca2+ channels are complex oligomeric proteins that are heavily regulated by many distinct signal transduction mechanisms, much less is known about the auxiliary subunit structure for T-type Ca2+ channels.50 The electrophysiological properties of T-type α1-subunits expressed alone are very similar to those observed in native channels, suggesting that T-type Ca2+ channels do not require auxiliary subunits, as do L-type Ca2+ channels, for proper function. Consistent with this, antisense depletion of CaVβ-subunits has no effect on ICa,T (in neurons),51,52 and coexpression of cloned CaVβ-subunits has no major effect on ICa,T in heterologous expression systems.53 CaVγ-subunits have been found to have little or no effect on CaV3.3 currents54; however, they have been found to accelerate inactivation and negatively shift steady state inactivation of the CaV3.1 current.55 Coexpression of α2δ-subunits with α1G doubled the size of ICa,T, possibly through increased expression of α1-α2δ complexes at the plasma membrane.53,56 T-type Ca2+ channels have been shown to interact with, and be modulated by, other proteins such as Kelch-like 1 protein57 and caveolin-3.58 In summary, available data indicate that CaV3 α1-subunits can interact with some auxiliary subunits, but additional studies are needed to determine the physiological role for these interactions, particularly in the heart. The biophysical properties of T-type Ca2+ channels in heart are distinct from those of L-type Ca2+ channels.59 For example, T-type Ca2+ channels activate and inactivate at lower (more negative) membrane potentials with thresholds for activation observed at about −70 mV while inactivation begins to occur at approximately −90 mV50 (see Figure 2-2). T-type Ca2+ channels also show much faster entry and exit from inactivation50,60 that is independent of Ca2+. An analysis of the steady state activation and inactivation curves reveals that T-type Ca2+ channels have relatively large steady state window currents at membrane voltages between −80 and −40 mV,61 which are expected to facilitate depolarization in myocytes of the SAN and the conducting system, thereby promoting spontaneous action potential firing. Although the permeation properties of T-type Ca2+ channels have been challenging to quantify, the single-channel conductance is reported to be 2.5-fold smaller than that of L-type Ca2+ channels (when Ba2+ is the charge carrier).62
Calcium Channels in the Heart
L-type Ca2+ Channels
Molecular Composition
Biophysical Properties of ICa,L
ICa,L Inactivation
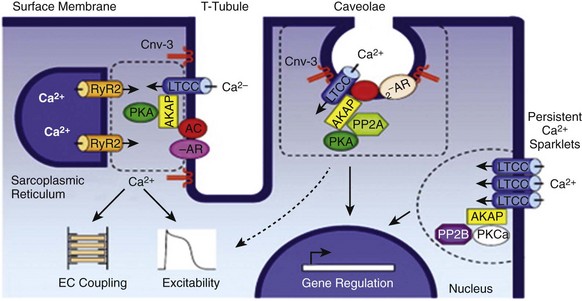
Organization and Localization of LTCCs
T-type Ca2+ Channels
Molecular Composition and Biophysical Properties
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Calcium Channels in the Heart
