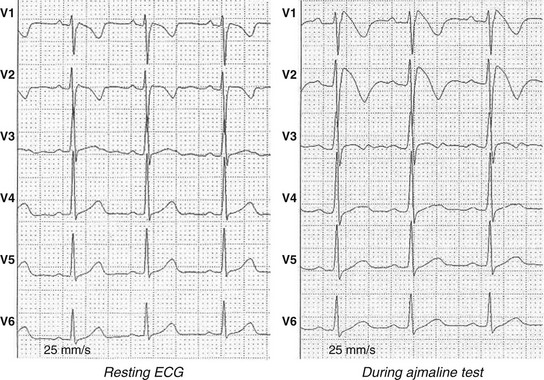
92 The year 2012 marked the 20th anniversary of publication of the manuscript, “Right Bundle Branch Block, Persistent ST Segment Elevation and Sudden Cardiac Death: A Distinct Clinical and Electrocardiographic Syndrome,” in Journal of the American College of Cardiology.1 This publication described eight individuals with a common phenotype: All had a structurally normal heart and had been resuscitated from sudden cardiac death caused by documented ventricular fibrillation. All had a characteristic ST segment elevation in the right precordial leads (Figure 92-1). Although it started as a sort of scientific curiosity, 20 years later this syndrome, now known as Brugada syndrome (BrS), is recognized as a major disease that integrates into a continuum of previous syndromes like idiopathic ventricular fibrillation, sudden unexplained death syndrome, some forms of sudden infant death syndrome, and overlapping syndromes with long or short QT intervals by tracing the origins of these different syndromes through the same pathophysiological background: alteration of ionic currents leading to ST elevation and ventricular arrhythmias. At least 13 different genetic variants of the syndrome are known nowadays, with more than 300 mutations reported. The extremely wide genetic heterogeneity of inherited arrhythmia disorders, along with the sometimes almost impossible genotype-to-phenotype correlations, the overlapping electrophysiological manifestations, and the difficulties associated with diagnosis and assessment of prognosis, makes this new field of genetic rhythmology a fascinating one. In this chapter we review present knowledge, progress that has been made, and future research directions for Brugada syndrome. Figure 92-1 Brugada Electrocardiogram (ECG) Patterns The diagnosis of BrS is based on clinical and electrocardiographic features. Some patients present with syncope or (aborted) sudden cardiac death caused by malignant ventricular arrhythmias; however, others are completely asymptomatic, and no apparent structural heart disease can be found. The hallmark of BrS is the transient or persistent appearance of typical electrocardiographic (ECG) changes in the right precordial leads. The second Brugada Syndrome Consensus Report of 2005 (endorsed by the Heart Rhythm Society and the European Heart Rhythm Association)2 stated the current recommendations regarding diagnostic criteria. Three different ECG patterns (see Figure 92-1), all featuring ST segment elevation in the right precordial leads, have been recognized: Type I is the only pattern that is diagnostic for BrS. It consists of a coved-type ST segment elevation greater than 2 mm, followed by a descending negative T wave in at least one right precordial lead (V1 to V3). Type II and type III are saddleback-shaped patterns, with high initial augmentation followed by an ST elevation greater than 2 mm for type II and less than 2 mm for type III. Both patterns are suggestive of but not diagnostic for BrS. Whenever a large number of baseline ECGs were available during follow-up, the diagnostic pattern could be documented in only approximately 25% of the tracings. Almost every individual with a type I ECG will show normalization of the ECG during follow-up. Because the presence of the spontaneous coved-type (type I) ECG pattern is a useful predictor of future arrhythmic events in asymptomatic patients, this variation of the ECG pattern is of great clinical importance. The full stomach test was proposed as a tool in diagnosing BrS3. Here, ST segment changes appear to be provoked by an enhanced vagal tone. Adrenergic stimulation decreases ST segment elevation, whereas vagal stimulation increases it. Obviously, it is important to exclude other causes of ST segment elevation before making the diagnosis of BrS. These other causes are shown in Table 92-1. The class IC antiarrhythmic drug is the main test for unmasking concealed forms.4 The diagnostic coved-type BrS ECG pattern could be elicited by intravenous administration of ajmaline, flecainide, or procainamide (Figure 92-2). On the basis of the results of comparative studies5 and clinical experience, ajmaline, at a dose of 1 mg/kg, is the best drug. Table 92-1 Acquired Brugada Syndrome: Differential Diagnosis of ST Segment Elevation in Electrocardiogram Leads V1 and V2 Brugada syndrome (BrS) is a channelopathy that induces an electrical dysfunction in channels that participate in generation of the cardiac AP. Experimental and clinical data have elucidated the cellular and molecular bases for the ECG morphology and arrhythmogenesis of BrS.4 Two mechanisms are believed to explain the ST segment elevation seen in the right precordial leads: First, a disequilibrium between INa and Ito that affects preferentially the right ventricular myocardium, generating transmural dispersion of repolarization and the substrate for arrhythmias. It has been suggested that the embryologic origin of the right ventricle (neural crest cells) differs from that of the left ventricle, and this fact predisposes the right ventricle to arrhythmias in adulthood.5 The cellular basis for this phenomenon is thought to be the result of a loss-of-function Na+ channel that differentially alters AP morphology in epicardial versus endocardial cells. The fast transient outward K+ current Ito is most prominent in epicardial cells of the right ventricle. This K+ current is quickly activated by membrane depolarization. It opposes and exceeds the depolarizing effect of Na+ inward flux during the early phase of the AP plateau, resulting in a pronounced AP notch and, in combination with depolarizing Ca2+ currents, in a “spike-and-dome” morphology. Consequently, Na+ current reduction leads to an outward shift of the net transmembrane current in epicardial cells, finally resulting in premature repolarization and significant AP shortening. In contrast, endocardial cells display a much smaller Ito, and consequently, Na+ current reduction would not significantly affect AP morphology and duration. The transmural homogeneity of the cellular membrane voltage finally causes ST segment elevation.6 The second mechanism that accounts for the ST segment elevation seen in the right precordial leads is conduction slowing in the right ventricular outflow tract (RVOT), leading to ST segment elevation in the right precordial leads.7 Brugada syndrome (BrS) is a familial disease with an autosomal dominant pattern of inheritance. To date, more than 250 mutations in 13 genes have been published (Table 92-2). The first gene associated with BrS was SCN5A, which encodes the α-subunit of the cardiac sodium channel.8 The SCN5A gene is responsible for phase 0 of the cardiac action potential. Mutations in SCN5A result in loss-of-function of the sodium channel. In all, 20% to 25% of patients with BrS have a mutation in the SCN5A gene, classified as BrS type 1.9 Other mutations are seen in SCN1B (sodium channel β1-subunit), which modifies Nav1.5, thus increasing INa,10 and in SCN3B (sodium channel β3-subunit), which alters Nav1.5 trafficking, thereby decreasing INa.11 Another gene reported as responsible for BrS is GPD1-L. Mutations in GPD1-L reduce both the surface membrane expression and the inward sodium current.12 Kattygnarath et al. published a study supporting that RANGRF (MOG1 protein) can impair the trafficking of Nav1.5 to the membrane, leading to INa reduction and clinical manifestations of BrS.13 Apart from sodium channels, several potassium channels have also been related to BrS. The first one described was KCNE3, which codifies the MiRP2 protein (β-subunit that regulates the potassium channel Ito) and modulates some potassium currents in the heart.14 Another gene associated with BrS is KCNJ8, which was previously related to early repolarization syndrome (ERS).15 KCNJ8 was described as a novel J wave syndrome susceptibility gene and a marker of gain-of-function in the cardiac K(ATP) Kir6.1 channel.16 In 2011, Giudicessi et al. provided the first molecular and functional evidence implicating novel KCND3 gain-of-function mutations (Kir4.3 protein) in the pathogenesis and phenotypic expression of BrS, with enhanced Ito current gradient within the right ventricle, where KCND3 gene expression is the highest.17 Also in 2011, novel variants of KCNE5 appeared to cause gain-of-function effects on Ito. The KCNE5 gene is located in the X chromosome and encodes an auxiliary β-subunit for K channels.18 BrS was also associated with HCN4, which codifies for the HCN4 channel or the If channel (hyperpolarization-activated cyclic nucleotide–gated potassium channel 4). Its mutations also predispose to inherited sick sinus syndrome (SSS) and to long QT syndrome (LQTS) associated with bradycardia.19 Calcium channels have also been associated with BrS. Mutations in the CACNA1C gene are responsible for a defective unit of the type L calcium channel. Mutation of the CACNB2B gene leads to a defect in the L-type calcium channel. Both induce loss of channel function.20 In 2010 the CACNA2D1 gene was reported as responsible for BrS. The α2/δ-subunit of voltage-dependent calcium channels regulates current density and activation/inactivation kinetics of the calcium channel.21 In recent years, several genetic and environmental modulators of the phenotype have been described. It is well known that environment may play a role in the predisposition to arrhythmias in patients with Brugada syndrome. The identification of several triggering factors of the Brugada ECG pattern and that of sudden cardiac death (SCD)—fever, cocaine, electrolyte disturbances, class I antiarrhythmic medications, and several other noncardiac medications,22 some of them with a genetic predisposition—has resulted in the need to adopt preventive measures in patients with the diagnostic ECG pattern.23 In addition, incomplete penetrance of the disease, as well as its variable expressivity, has brought into question the role of additional genetic factors in the final phenotype. Single-nucleotide polymorphisms (SNPs) became one of the first players to be studied in defining this variability. The SCN5A polymorphism H558R is present in 20% of the population. This polymorphism has been shown to partially restore the sodium current impaired by other simultaneous BrS-causing mutations in SCN5A.24 Thus, this common variant is a genetic modulator of BrS among carriers of an SCN5A mutation, in whom the presence of the less common allele makes BrS less severe.25 Genetics variants in the SCN5A promoter region may also play a pathophysiological role in BrS. A haplotype of six polymorphisms in the SCN5A promoter has been identified and has been functionally linked to reduced expression of the sodium current in the Japanese population.26 Other studies have shown the role of double mutants in causing a more severe phenotype.27,28 The role of the genetic mutation in risk stratification has yet to be clearly defined. Recent data suggest that the type of genetic mutation can serve as a tool for risk stratification in BrS. In this study, patients and relatives with a truncated protein had a more severe phenotype and more severe conduction disorders. Although this provides proof of the concept that some mutations appear to confer a worse prognosis, use of these data in the clinical setting is not yet sufficient to alter clinical decisions.29 The early repolarization syndrome (ERS) is a common electrocardiographic variant characterized by J point elevation, ST segment elevation with upper concavity, and prominent T waves in at least two contiguous leads.30 Despite the overlap degree between ERS and BrS remains undetermined, ERS and BrS share cellular, ionic, and ECG similarities (appearance of J waves), representing parts of a phenotypic spectrum called J wave syndromes.31 In the last 5 years, it has been published patients with BrS and ERS.32 ERS has been linked to mutations in CACNA1C, CACNB2, CACNA2D1, and KCNJ8.33 Lev-Lenègre syndrome (also called progressive cardiac conduction disease [PCCD]) is a rare entity characterized by disruption of the conduction system through the His-Purkinje system, associated with syncope and even SCD. The presence of PCCD among BrS families is not uncommon, as both conditions result from a reduction in the sodium current, which has been described as a different expression of the same genetic phenotype. The first mutations associated with PCCD were described with the SCN5A gene34,35 and its β1-subunit.10 Sick sinus syndrome (SSS) is characterized by persistent inappropriate sinus bradycardia, sinus arrest, atrial standstill, and tachycardia–bradycardia syndrome, all of which are associated with dysfunction of the sinoatrial node (SAN). Patients may exhibit varied symptoms including syncope and even SCD. The course of SSS can be intermittent and unpredictable, according to the severity of the underlying heart disease.36 So far, both autosomal recessive and dominant forms have been described. In 2003 the association between SCN5A mutations and congenital SSS was reported.37 In 2005 a novel SCN5A mutation was identified in patients presenting with both SSS and BrS,38 showing that in the same family, both diseases may be related to the expression of a loss-of-function mutation in INa. Atrial fibrillation (AF) is the most common atrial arrhythmia found in BrS.39 It is of extreme clinical importance to realize that atrial fibrillation can be the first manifestation of Brugada syndrome. Administration of antiarrhythmic drugs in these cases can lead to ventricular fibrillation and sudden death. Brugada syndrome should be excluded by drug challenge in young individuals with atrial flutter or fibrillation and a normal heart and a normal ECG. Approximately 15% to 20% of patients with BrS develop supraventricular arrhythmias.40 Some studies have reported prolongation of atria His and the His ventricular (HV) interval; these changes occur principally in patients with SCN5A mutations41 and are consistent with decreased excitability in the conduction system secondary to loss-of-function of sodium channel activity.42
Brugada Syndrome 1992–2012
Twenty Years of Scientific Progress
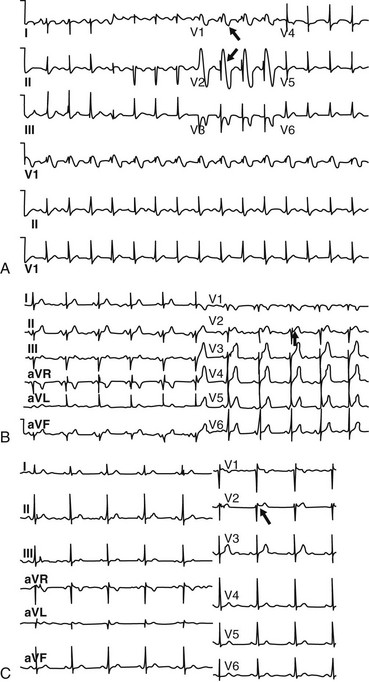
A, A diagnostic coved-type (type I) Brugada ECG pattern documented in a 9-year-old girl who presented with syncope and a positive family history of Brugada syndrome. Note the pattern resembling a right bundle branch block (arrows) in leads V1 and V2, with typical ST elevation. B, Baseline ECG of a 58-year-old asymptomatic man with a positive family history of Brugada syndrome. This is an example of a type II saddleback Brugada ECG pattern. Genetic analysis revealed a mutation in the SCN5A gene. Note the saddleback-shaped patterns, with high initial augmentation followed by an ST elevation greater than 2 mm in lead V2. C, Example of a baseline type III saddleback Brugada ECG pattern (arrow) documented in a 61-year-old asymptomatic man who was diagnosed on the basis of a positive result on class IC antiarrhythmic drug testing.
The Electrocardiogram

Mechanisms, Biophysics, and Molecular Genetics
Molecular Mechanisms
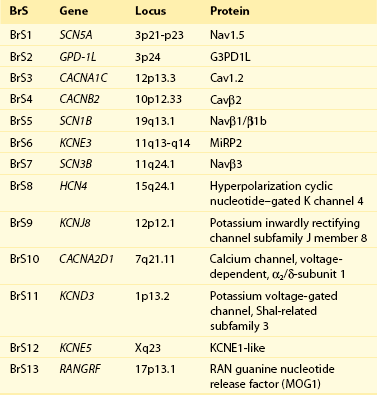
Genetics
Genetic and Environmental Modulators
Brugada Syndrome and Overlapping Syndromes
Early Repolarization Syndrome
Lev-Lenègre Syndrome
Sick Sinus Syndrome
Atrial Fibrillation
Brugada Syndrome 1992–2012: Twenty Years of Scientific Progress