Bronchiectasis
Bronchiectasis (broncos, airways; ectasia, dilatation) is a morphologic term used to describe abnormal, irreversibly dilated and thick-walled bronchi. This is an anatomic definition that evolved from Laennec’s original description in 1819 of ectatic bronchi in pathologic specimens. Bronchiectasis represents the end stage of a variety of pathologic processes that cause destruction of the bronchial wall and its surrounding supporting tissues. Etiologies include prior lung infection, systemic inflammatory disorders, and genetic disorders of host defense, however, bronchiectasis is considered to be idiopathic in up to half of the affected individuals. The clinical manifestations include chronic cough and copious mucopurulent expectoration.1 Bronchiectasis shares many features with chronic bronchitis, including inflamed and easily collapsible airways, airflow obstruction on spirometry, and frequent exacerbations.
PREVALENCE
Bronchiectasis was a common disabling and fatal condition in the pre-antibiotic era and remains more common in medically underserved regions of the world. Overall, it is an important cause of suppurative lung disease with a significant impact on the quality of life of affected individuals and on the health system as patients utilize many medical care resources including frequent clinic visits, hospitalizations, diagnostic imaging such as high-resolution computed tomography scan (HRCT) of the chest and parenteral antibiotics.2 In the United States, the overall prevalence has been estimated to be 52 per 100,000, but varies by age. In persons aged 18 to 34 years the prevalence is approximately 4.2 per 100,000 but in those who are 75 years old or it is estimated to be greater than 272 per 100,000. There are an estimated 110,000 affected individuals in the United States.3 In most series, 60% of affected individuals are women. The incidence is higher in some ethnic groups living in isolated regions including the native peoples in Alaska, Maori populations in New Zealand and the Pacific, and Aboriginal groups in Central Australia. In North America and Europe, improved health care has decreased the incidence, thus bronchiectasis due to cystic fibrosis (CF) and other genetic diseases now significantly contribute to the fraction of affected adults.
PATHOPHYSIOLOGY
The pathogenesis of bronchiectasis is not known in many cases, and in others may vary with etiology, so that the pathophysiology often remains descriptive. Gross pathology reflects chronic changes so that initial changes of injury proposed to lead to initial airway obstruction are not often observed. The abnormal bronchial dilatation in bronchiectasis principally affects the medium-sized bronchi, but typically extends to the distal bronchi and bronchioles. On gross examination of surgically resected or autopsied lungs, the affected bronchi and bronchioles are so prominent as to be visible all the way to the pleural surface. These dilated and ectatic bronchi are commonly filled with purulent secretions. The affected bronchi show transmural inflammation, mucosal edema, cratering, ulceration, and neovascularization. The bronchial epithelium may show a polypoidal appearance due to underlying granuloma formation and lymphoid aggregates, ridging due to bronchial smooth muscle hypertrophy, and pitting due to the dilated bronchial mucus glands. Severe cases may show denudation of epithelial lining, with destruction of underlying elastic laminae, smooth muscle, and cartilage with fibrotic changes replacing these structures. Dilated and tortuous bronchial arteries may be seen secondary to the development of extensive bronchial-pulmonary anastomoses.
Microscopically, bronchiectasis is associated with airway epithelial remodeling characterized by mucus cell metaplasia, and decrease in ciliated cells. In other regions, cuboidal and squamous metaplasia predominate. Intense infiltration of the bronchial wall with neutrophils, lymphocytes, and monocytes is common. Hypertrophy of bronchial glands, and lymphoid hyperplasia are also seen.
Various explanations have been advanced for the phenomenon of bronchiectasis after bronchial obstruction. Following bronchial obstruction, airways proximal to the collapse are exposed to strong dilating forces caused by the difference in the atmospheric pressure in the bronchi and the negative pressure in the pleural space. Over time, these forces acting on weakened, inflamed airways may result in permanent and pathologic airway dilatation. The presence of surrounding lung fibrosis, atelectasis, and loss of lung volume leading to regional increases in local retractile lung forces may also play a role. Animal experiments suggest that obstruction may facilitate the development of bronchiectasis by interfering with bronchial clearance and promoting bacterial infection, bronchial wall inflammation, and weakening.
It has long been recognized that the pathologic changes are associated with chronic bacterial infection, independent of the initial cause of bronchiectasis. The concept of the “vicious cycle” of recurrent infectious and inflammatory insults proposed by Peter Cole et al.,4 37 years ago has largely been accepted. The cycle is initiated by an infection and an airway insult in a host at genetic risk. This vicious cycle theory proposed that chronic bacterial endobronchial infection and inflammation damage or destroy mucociliary defenses, leading to secretion stasis, which in turn propagates furthers bacterial infection, and increases airway inflammation and bronchial dilatation. Specific primary defects in innate host defense (e.g., IgG deficiency) have been identified as causative.5 Furthermore, acute bacterial infection and chronic biofilm formation in the airways alone are not sufficient to produce true bronchiectasis. Impaired airway epithelial cell function, immune response, or other systemic inflammatory conditions resulting in insufficient airway clearance are additionally required. Once injury ensues, the appearance of Pseudomonas aeruginosa in the respiratory tract of bronchiectasis patients on a chronic or recurring basis has been associated with worsening airway clearance and airway obstruction, resulting in impaired health-related quality of life (HQOL) and worsened lung function.6 This may be due to the ability of this organism to release virulent exotoxins, form biofilms on tissue surfaces, and easily develop hypermutable P. aeruginosa strains resistant to antibiotics, all factors perpetuating and propagating bronchial damage.
Specificity biochemical and molecular markers of non-CF bronchiectasis are not established and robust animal models of bronchiectasis have not been developed. Early biochemical changes are proposed related to infection and reflect a stereotyped host immune response. Later in disease, neutrophils, macrophages, and monocytes, along with their products are abundant. At both stages, cytokines such as tumor necrosis factor-a (TNF-a) and interleukin-8 may be elevated, providing a sustained signal for inflammatory cell recruitment.7 Neutrophil- and macrophage-derived elastases and proteases isolated in large quantities in sputum have been proposed to contribute to airway injury and subsequent chronic bronchiectasis pathology.8
CLINICAL FEATURES
The classic clinical manifestations of bronchiectasis are daily cough and mucopurulent sputum production. Cough is invariably present and often may be the only symptom for years. Purulent, tenacious sputum production, frequently worse in the morning (having accumulated during recumbency in sleep) is present in most patients. Sputum production may be intermittent, being affected by recurrent infections, bronchial plugging, and antibiotic therapy. “Dry bronchiectasis” presenting as cough, minimal sputum expectoration, and/or hemoptysis is occasionally described. Hemoptysis may be seen in 40% to 70% of patients and may vary from blood streaks to large clots. Increasing cough, dyspnea, and volume and darkening of sputum color, fever, hemoptysis, and chest pain are hallmarks of acute exacerbations. Often patients give a history of recurrent chest infections, although single episodes of severe pneumonia, tuberculosis, or pertussis with secondary pneumonia may also result in bronchiectasis.
On physical examination chest auscultation usually reveals findings of early and midinspiratory crackles as well as diffuse rhonchi and prolonged expiration. Bronchial breath sounds may be heard in severe cases or patients with a complicating pneumonia. Digital clubbing and hypertrophic pulmonary osteoarthropathy, although common in the pre-antibiotic era, are rarely seen now. In advanced cases, there may be evidence of respiratory insufficiency and cor pulmonale.9
PREDISPOSING OR ASSOCIATED CONDITIONS
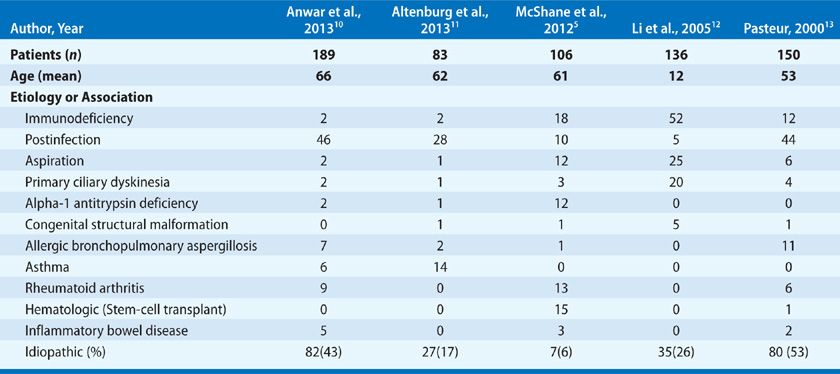
Previously bronchial damage secondary to childhood respiratory tract infections such as pneumonia, pertussis, complicated measles, and tuberculosis were implicated as common causes of bronchiectasis. However, with the early use of antibiotics and childhood immunizations, the focus has shifted from postinfectious to intrinsic host defense causes. Often regarded as a condition in which extensive investigation is unlikely to yield treatable causes, recent studies have shown results to the contrary (Table 53-1). Most series from referral centers identified an association or contributing cause in approximately 50% of patients. Careful investigation and the identification of the genetic basis for immune and other disorders of innate airway defense have reduced the number of idiopathic cases and increased the number of individuals that may considered for specific therapy. The overall impact of these efforts is a bimodal distribution in the age of adult patients with bronchiectasis composed of those with genetic causes first presenting in childhood and an elderly population, typically composing the idiopathic group.
 INFECTION
INFECTION
A number of pulmonary infections have been associated with the development of bronchiectasis. Complicating secondary infections with adenovirus, herpesvirus, and bacteria such as Staphylococcus aureus, Klebsiella pneumoniae, and Pseudomonas aeruginosa contribute to the severity of a necrotizing bronchopneumonia. Streptococcus pneumoniae, Haemophilus influenzae, and Moraxella infections typically do not cause bronchiectasis, but may be chronically present within bronchiectatic airways. Necrotizing pneumonias secondary to chronic aspiration or bronchial obstruction are often complicated by parenchymal destruction and bronchiectasis. Tuberculosis can result in bronchiectasis by several mechanisms. Bronchiectasis may be a consequence of tuberculous bronchitis, postobstructive bronchial damage secondary to posttuberculous bronchial wall stenosis, and extraluminal bronchial obstruction by enlarged tuberculous lymph nodes.
The association of nontuberculous Mycobacterium (NTM) with bronchiectasis is well documented. CT scan of the chest in such cases is relatively specific, showing small irregular nodules in the middle lobe or lingula, but other parts of the lung may be affected. While traditionally considered a secondary pathogen in an abnormal host or a colonizer in damaged lungs (bullous emphysema, cavitary lung disease) it is now recognized that NTM can cause bronchiectasis in apparently normal hosts. A recent study of individuals with idiopathic bronchiectasis infected with NTM demonstrated shared features with primary ciliary dyskinesia (PCD) (slow cilia beat frequency, low nasal nitric oxide) suggesting the presence of either an underlying genetic or acquired defect.14 One phenotype for NTM-associated bronchiectasis seems to involve predominantly slender white women 50 to 70 years old with kyphoscoliosis. An underlying primary lung disease or immune defect has not been identified in this subpopulation.15
 BRONCHIAL OBSTRUCTION
BRONCHIAL OBSTRUCTION
Localized bronchiectasis may also been seen in middle-lobe syndrome, and is usually caused by intraluminal or extraluminal obstruction secondary to tumor, enlarged lymph nodes, or abnormalities of bronchial structure and branching. Endobronchial adenomas, fibromas, chondromas, and lower respiratory tract papillomatosis causing partial airway obstruction and bronchiectasis have been described.
 ASPIRATION/INHALATION AIRWAY INJURY
ASPIRATION/INHALATION AIRWAY INJURY
Aspiration or inhalation of foreign matter, such as noxious fumes or particulates into the airways, may result in bronchiectasis. This may involve aspiration of oropharyngeal secretions containing microaerophilic and anaerobic bacteria, leading to a necrotizing pneumonia. Refluxed material from the esophagus or stomach containing food particles, gastric, biliary, and pancreatic secretions, and gut microbes may enter and damage airways, especially if the aspiration events are large and repeated. Depressed sensorium (stroke, alcohol and drug use, seizure, postanesthetic), neuronal or spinal cord dysfunction (amyotrophic lateral sclerosis, multiple sclerosis, syringomyelia), defective laryngeal function (postsurgery, postirradiation), esophageal disorders (dysmotility, achalasia, tracheoesophageal fistula), and gastric disorders (gastric outlet obstruction) influence the likelihood and frequency of aspiration. Bronchiectasis may present years after foreign body aspiration (aspiration is often unrecognized), although bronchiectasis has been seen to occur in animals as soon as 2 to 8 weeks after experimental foreign body introduction into the bronchial tree. GERD is the most common condition in this category contributing to the risk of bronchiectasis CCDC.
 CYSTIC FIBROSIS
CYSTIC FIBROSIS
CF is a common cause of bronchiectasis in the United States and other developed countries (see Chapter 50). This is an autosomal recessive, monogenic disorder that presents most commonly in childhood as a multisystem disease. However, 3% to 7% of patients with CF are diagnosed in adulthood, and due to improvements in therapy, there are now more adults over 18 years old with CF than younger individuals. CF is caused by a genetic deficiency in the Cystic fibrosis transmembrane conductance regulator (CFTR). Approximately 2000 mutations in the CF gene have been identified. Clues suggesting CF as a cause of bronchiectasis include upper lobe radiographic involvement and sputum cultures showing mucoid P. aeruginosa or S. aureus. The diagnosis of CF rests on a combination of clinical criteria accompanied by sweat chloride values above 40 to 60 mmol/L. However, intermediate or normal sweat chloride values may be seen in patients with clinical manifestations of CF and genetically confirmed CF. Screening for other mutations in the CFTR gene may be necessary in these circumstances. Measurement of the electrical potential difference across the nasal epithelium, available in specialized centers, is sometimes used to corroborate the diagnosis.
 PRIMARY CILIARY DYSKINESIA
PRIMARY CILIARY DYSKINESIA
PCD is a genetically heterogeneous syndrome caused by defect in motile cilia. The true prevalence is unknown but estimated to affect 1:20,000 to 1:100,000 people.16 The tissue-specific location of cells with motile cilia reflects the clinical features including chronic otitis media, rhinosinusitis, bronchiectasis, infertility, and laterality defects including situs inversus. PCD has an autosomal-recessive inheritance pattern and has been ascribed to mutations in over 30 genes to date that are estimated to account for approximately 60% of all cases. The involvement of many genes in this syndrome is consistent with the knowledge that over 2000 proteins are involved with cilia assembly, structure and function. The ciliary axoneme contains nine outer and two inner pairs of microtubules that are connected to dynein motor proteins forming characteristic structures of motor complexes called inner and outer dynein arms that can be observed by transmission electron microscopy of preparations of cilia from respiratory epithelial cells. Known causative genes in PCD encode proteins for dynein motors (e.g., DNAI1, DNAI2, DNAH5, DNAH11), cilia motor regulation and structural assembly (e.g., RSPH4 A, RSPH9, CCDC39, CCDC40) and motor complex preassembly (e.g., DNAAF1, DNAAF2, HEATR2).16
In 1933 Kartagener described the PCD syndrome, as the triad of situs inversus, bronchiectasis, and either nasal polyps or recurrent sinusitis, while the description by Afzelius in 1976 of the defects in the ultrastructure of ciliary dynein arms revealed the basis of this condition.17,18Thus, clinical findings include, respiratory distress in neonates, recurrent respiratory tract infections, bronchiectasis, situs inversus, infertility, and heterotaxy in approximately 50%. Laterality defects are the result of motile cilia defects in the embryonic node, a midline structure transiently present during early development that contains cilia. Directional movement of fluid in the node activates downstream programs that establish left and right sidedness of organs. In the absence of flow, situs solitus (normal left-right), situs inversus (fully reversed right-left, functional organs), or intermediate states may occur resulting in cardiac defects. Thus, individuals with congenital heart disease may also have cilia dysfunction, complicating exacerbation of pulmonary disease in heart failure.19 Hydrocephalus due to dysfunction of the motile cilia of the brain ventricles is exceedingly rare.
In a study of 94 patients from 68 families, Noone et al.20 showed that cough was seen in 100% of patients, bronchiectasis (98%), sinusitis (47%), otitis media (92%), and situs inversus (46%). Although most patients with PCD are identified in childhood, this disorder may not be accurately diagnosed until adulthood. Like CF, bronchiectasis occurs in children and is progressive, however in contrast to CF, lung disease is not as severe and lifespan is usually normal.
Accurate testing for PCD is technically demanding and should be performed in specialized centers.21 Nasal nitric oxide (NO) is emerging as the most sensitive screening test, with levels of NO being characteristically low, a feature that can be shared with CF.20 Once CF is excluded (e.g., by sweat test), then nasal NO together with clinical features provides high specificity for diagnosis. Ciliated epithelial cells obtained from the inferior or middle turbinate using a sterile cytology brush may be studied for ciliary beat pattern and frequency using digital high-speed video imaging. This requires experienced observers. Abnormalities in ciliary beat have been correlated to ultrastructural defects, but normal ciliary motion cannot fully exclude PCD since some mutations are associated with near normal beat frequencies. Axonemal structure of respiratory cilia may be visualized by transmission electron microscopy and defects in dynein arms, peripheral and central tubules, radial spokes, and basal bodies may be seen. These studies are technically challenging and no structural abnormalities may be found in cases of PCD,22 particularly when mutations involve regulatory proteins. Genetic testing is available in some research centers.
 COPD AND BRONCHIECTASIS
COPD AND BRONCHIECTASIS
Cigarette smoking causes COPD but is probably not an etiology for bronchiectasis although there is overlap in clinical characteristics that have prognostic and management implications. The phenotype includes frequent exacerbations requiring medical attention in an urgent care setting,23 HRCT scans that have both emphysema and bronchiectasis portend a reduced prognosis,24 obstructive impairment on pulmonary function, and sputum microbiology that contains P. aeruginosa.6,25,26
 ALPHA-1 ANTITRYPSIN DEFICIENCY (AATD)
ALPHA-1 ANTITRYPSIN DEFICIENCY (AATD)
Unimpeded neutrophil elastase contributes to the alveolar destruction of emphysema in AATD. Abundant elastin in airways could also be subject to destruction. There may be a spectrum of disease with heterogeneous lung conditions rather than simply pure emphysema in patients with AATD. Small case series have identified bronchiectatic changes in patients with AATD. Cuvelier et al. measured AAT alleles in patients with known bronchiectasis and healthy blood donors. They did not find any significant differences in AAT alleles between patients with bronchiectasis and control individuals except in those patients with both emphysema and bronchiectasis. There were more abnormal alpha-1 alleles in those patients with coexisting emphysema and bronchiectasis. They concluded that bronchiectasis might be a consequence of emphysema.27 In a study of 74 patients by Parr et al. with AATD (PiZ phenotype), 70 out of 74 were found to have bronchiectasis on HRCT scan. They defined clinically significant bronchiectasis as patients with regular sputum production and HRCT findings of bronchiectasis affecting four or more lobes.28 Fifty-seven patients had bronchiectasis in four or more bronchopulmonary segments. Twenty (27%) met the criteria for clinically significant bronchiectasis. In general, those with more severe bronchiectasis had more severe emphysema. Whether there is a common pathway that contributes to both emphysema and bronchiectasis in patients with AATD or whether emphysema predisposes to bronchiectasis is still unknown.28
 ALLERGIC BRONCHOPULMONARY ASPERGILLOSIS
ALLERGIC BRONCHOPULMONARY ASPERGILLOSIS
Allergic bronchopulmonary aspergillosis (ABPA) is a hypersensitivity lung disease caused by the ubiquitous fungus Aspergillus fumigatus and usually occurs as a complication of persistent asthma or CF. The excessive mucus production and impaired mucociliary clearance in these conditions allow the inhaled conidia of Aspergillus to persist and germinate, releasing exoproteases and other fungal products that further compromise clearance, breach epithelium, and activate immune responses. ABPA is characterized by a marked local and systemic eosinophilia, an elevated level of A. fumigatus—specific IgG and IgE antibodies, as well as a nonspecific elevation of total IgE. Clinically, ABPA manifests as difficult-to-control or recurring episodes of asthma, pulmonary infiltrates, and central bronchiectasis that may progress to fibrosis. Criteria have been established for the diagnosis of ABPA in the non-CF as well as the CF population.29
 INFLAMMATORY DISORDERS
INFLAMMATORY DISORDERS
Inflammatory and fibrotic processes affecting large and small airways may be seen in several rheumatic diseases and autoimmune states. Significantly higher frequencies of bronchiectasis (20%–35%) have been found in rheumatoid arthritis (RA) patients undergoing HRCT, both in symptomatic (30%) and asymptomatic (8%) patients, and was independent of smoking status. Bronchiectasis may precede or follow the development of RA, and the coexistence of both conditions is considered to portend a reduced survival.30 Sjögren syndrome may also be complicated by bronchiectasis presumed to be secondary to the effects of inspissated bronchial secretions causing atelectasis and bronchial wall destruction.31 Relapsing polychondritis may be complicated by bronchiectasis in regions of recurring pneumonia as well as regions free of infection. It is not clear whether the chondritis itself or the recurrent infections predispose to bronchiectasis. Inflammatory bowel disease such as chronic ulcerative colitis is associated with bronchiectasis. The pathogenesis remains unknown, although autoimmune and immune complex deposition theories have been proposed. This variant of bronchiectasis does not respond to colectomy and has been known to appear and progress after colectomy. Bronchiectasis seen in sarcoidosis is usually traction bronchiectasis secondary to parenchymal and peribronchial fibrosis. Endobronchial sarcoid may result in localized bronchiectasis secondary to obstruction, atelectasis, and bronchial wall destruction.32
 IMMUNE DEFICIENCIES
IMMUNE DEFICIENCIES
Bronchiectasis is associated with defects in both cellular and humoral immunity. Bronchiectasis is found in HIV and HTLV-1 infected individuals.33 It is also associated with acquired immunodeficiencies associated with stem-cell transplant and chemotherapy.5,12 Recurrent sinopulmonary infections and bronchiectasis are associated with defects in humoral immunity and hypogammaglobulinemia. Several forms of antibody deficiency have been linked with the development of bronchiectasis, including X-linked agammaglobulinemia, common variable immunodeficiency, IgA deficiency, and IgG subclass deficiency (usually IgG-G2 and IgG-G4).34 The issue of subclass deficiency (in the presence of normal or near-normal levels of total IgG) as a cause of bronchiectasis is controversial due to the wide range of values in normal individuals and the difficulties involved in accurately measuring these levels. An immunizing challenge with common humoral bacterial antigens, such as capsular polysaccharides of H. influenzae and S. pneumoniae followed by measurement of antibody titers 4 to 6 weeks later, may help establish the presence of such a deficiency. The lack of an antibody response is suggestive that humoral deficiency is present.35 Early diagnosis of these conditions and replacement with intravenous immunoglobulin significantly reduces infections and prevents bronchiectasis, although the efficacy of this treatment in patients with selective IgM, IgA, and IgG subclass deficiency remains controversial. Standard doses in adults of 300 mg/kg by intravenous infusion every 4 weeks have been proved to reduce rates and severity of respiratory infections, but higher doses of 600 mg/kg appear more efficacious in reducing respiratory exacerbations and preserving pulmonary function in some patients.36 Hyper-IgE syndrome is accompanied by recurrent lower respiratory infections leading to bronchiectasis and cystic lung destruction. Respiratory infection with Pseudomonas is a contributor to mortality.37
DIAGNOSIS OF BRONCHIECTASIS
The diagnosis of bronchiectasis is based on history, clinical features, and radiologic demonstration of bronchiectatic airways. The diagnostic evaluation in these patients is largely aimed at identifying potentially treatable underlying causes of bronchiectasis (Table 53-2). After confirming the diagnosis with chest imaging, CBC with differential for eosinophils, serum immunoglobulins IgG, IgA, and gM, sweat chloride or genetic CF testing, sputum culture for bacteria, mycobacteria, and fungi are productive starting tests. Subsequent testing will depend on clues from the history and likelihood of other conditions.



