Biology of the Vessel Wall: Introduction
Diverse pathologic processes contribute to common vascular diseases such as atherosclerosis and hypertension. During the past 2 decades, these pathologic events have been defined with increasing clarity at a cellular and molecular level, and strategies are emerging to treat these primary processes rather than simply treating the secondary manifestations of vascular disease. For this reason, understanding normal functions of vascular cells and how they are altered by various vascular insults has become essential for both basic investigators and clinicians caring for patients with peripheral vascular disease, coronary artery disease, and hypertension. This chapter introduces important concepts in vascular biology and emphasizes how fundamental aspects of vascular control are altered in common disease conditions.
The Endothelial Cell
Normal endothelial cell function is crucial to homeostasis in the vascular system. During the past 30 years, it has become apparent that diseases such as atherosclerosis are ultimately manifestations of endothelial dysfunction. Normally, the endothelium has three major roles: (1) it is a metabolically active secretory tissue; (2) it serves as an anticoagulant, antithrombotic surface; and (3) it provides a barrier to the indiscriminate passage of blood constituents into the arterial wall. The implications of these physiologic properties for vascular biology are considered separately.
Endothelial cells secrete vasoactive substances that play a major role in the control of vascular tone. These molecules include vasodilators such as prostacyclin, nitric oxide (NO), and endothelial-derived hyperpolarizing factors (EDHFs) such as hydrogen peroxide and eicosanoids.1-3 In addition, the endothelium produces vasoconstrictor substances, including endothelin4 and vasoconstrictor prostanoids.5
Endothelial cells also manufacture and secrete substances such as factor VIII antigen, von Willebrand factor, tissue factor, thrombomodulin, and tissue plasminogen activator (tPA), which are all involved in coagulation/fibrinolytic pathways. Structural components of the extracellular matrix (ECM) synthesized by these cells include collagen, elastin, glycosaminoglycans, and fibronectin.6,7 The composition of the ECM is dynamically modulated by matrix metalloproteinases (MMPs), enzymes that degrade matrix protein and participate in its remodeling. These enzymes are secreted by both endothelial and smooth muscle cells (SMCs).8,9 In addition, endothelial cells synthesize and secrete heparans and growth factors that regulate SMC proliferation.10-13 Finally, endothelial cells can clear and metabolically alter bloodborne and locally produced substances, including plasma lipids and lipoproteins,14 adenine nucleotides and nucleosides,15 serotonin, catecholamines, bradykinin, and angiotensin I.16
Endothelial cells are involved in the metabolism of plasma lipids in several ways. Lipoprotein lipase, an enzyme that hydrolyzes triglycerides into constituent fatty acids, is bound to the endothelial cell surface by heparan sulfates.17 The interaction of this enzyme with chylomicrons or very low-density lipoprotein particles results in the release of free fatty acids, which can then cross the subendothelial space to the underlying SMC or inflammatory cells in atherosclerosis. In addition, endothelial cells possess receptors for low-density lipoprotein (LDL),18 which regulate the transport and modification of LDL. Normally, LDL receptors are downregulated because receptor processing is inhibited in the nonproliferating monolayer.18 There are, however, two other pathways for uptake of LDL. First, LDL can be transported across the endothelium by an active process that is likely independent of plasmalemmal vesicles but may use paracellular gaps or fixed transendothelial channels.19 Second, modified, or oxidized, LDL can be taken up by scavenger LDL receptors,20 which include SRA, SR-BI, CD36 and the lectinlike oxidized LDL receptor-1.21,22 Endothelial cells also have the capacity to modify LDL,23 thus enhancing its uptake and ultimately leading to an increase in cholesterol esters in the vessel wall.
Recently, important new insights have emerged concerning the central role of metabolism in modulating not only the function of the endothelial cell itself but also the physiology and pathophysiology of the surrounding vascular and nonvascular parenchymal cells that are nurtured by the capillary networks. The metabolic state of the endothelial cell is determined to an important extent by the level of oxygen availability.24 Physical forces such as shear stress and the general metabolic and inflammatory milieu as reflected in levels of, for example, blood glucose and lipids as well as cytokines are also important in modulating the metabolic state and function of the endothelium as discussed subsequently.
Quiescent endothelial cells normally present an antithrombotic surface that inhibits platelet adhesion and coagulation. (For a more detailed discussion of thrombosis see Chap. 52.) Endothelial cells are, however, capable of synthesizing and secreting prothrombotic factors, especially when stimulated with cytokines or other inflammatory agents. The endothelium thus represents a functional antithrombotic, thrombolytic, or thrombotic balance. Potent anticoagulants elaborated by the endothelium include prostacyclin and NO (which inhibit platelet aggregation)25 antithrombin III,26 heparin-like molecules,27 and thrombomodulin (which activates endothelial protein C)28 and tPA. Procoagulant factors that can be produced by the endothelium include tissue factor,29 factor VIII, factor Va, and PAI-1 (plasminogen activator inhibitor 1) (Fig. 8–1). Conditions of injury or inflammation enhance the prothrombotic state of the endothelium by stimulating production of tissue factor and PAI-1. Of particular importance is tissue factor, which initiates the extrinsic coagulation pathway. The transcription and release of tissue factor is regulated by a myriad of proinflammatory, proatherogenic stimuli, and large amounts of tissue factor are associated with complex atherosclerotic lesions.30 There has also been substantial interest in the role of PAI-1 in vascular disease. PAI-1 levels are substantially elevated in humans with atherosclerosis and are even higher in the setting of acute coronary syndromes. Moreover, the metabolic syndrome, consisting of dyslipidemia, obesity, and insulin resistance, is associated with higher levels of PAI-1.31 Angiotensin II and thrombin likewise stimulate endothelial PAI-1 production, promoting thrombosis.32 Thus, under inflammatory conditions, endothelial cells can amplify the prothrombotic response. Not all factors controlling the expression of pro- and antithrombotic/fibrinolytic molecules are known, but it is clear that the endothelium functions as a major regulator of hemostasis.
Figure 8–1.
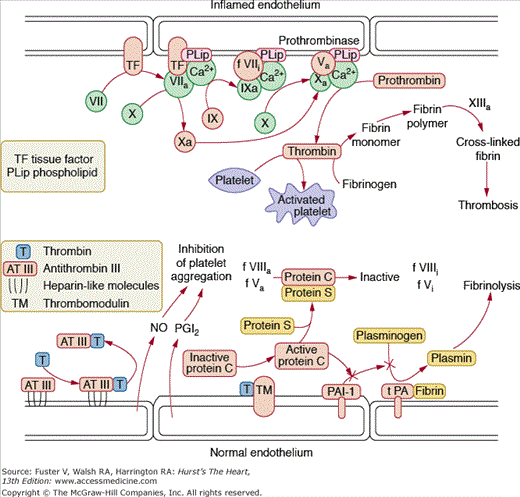
Pathways of thrombosis and thrombolysis. Under normal conditions, the endothelium is antithrombotic. Antithrombin III (AT III) binds thrombin and serves to clear thrombin from the circulation. Prostacyclin (prostaglandin I2 [PGI2]) inhibits platelet aggregation, and thrombomodulin (TM) activates protein C, which inhibits plasminogen activator inhibitor 1 (PAI-1) and interacts with protein S to inactivate activated factors V and VIII, thus limiting thrombosis. Because PAI-1 inhibits the tissue plasminogen activator (PA)—catalyzed conversion of plasminogen to plasmin, PAI-1 inhibition leads to accumulation of plasmin and fibrinolysis. With stimulation with inflammatory cytokines, there is increased expression of tissue factor on the endothelial cell surface. Tissue factor participates in the activation of factor X, which, in turn, promotes assembly of the prothrombinase complex, producing thrombin. Under these conditions, endothelial cells thus amplify the thrombotic response. Courtesy of Bernard Lassègue, PhD.
An essential role of the endothelium is regulation of permeability to macromolecules. The consequences of fluid and macromolecular transport vary depending on vessel size. In large vessels, these processes contribute to vessel nutrition and act as a selective barrier. In the microcirculation, endothelial permeability regulates delivery of nutrients to target organs and exchange of metabolic byproducts.
The two major mechanisms regulating endothelial barrier function involve modulation of intercellular contacts and transendothelial vesicular transport in caveolae. Two types of junctions regulate endothelial cell contact: adherens and tight junctions. Adherens junctions contain the protein vascular endothelial (VE) cadherin, which is essential for maintenance of interendothelial cell contacts. VE-cadherin associates with catenins, plakoglobulin, and the actin cytoskeleton to support cell adhesion. The catenins in turn bind to the actin cytoskeleton and modulate actin dynamics.33 Tight junctions are composed of occludins, claudins, and junctional adhesion molecule-1. Regulation of these interendothelial cell contacts is dynamic and important in modulation of new vessel growth, the extravasation of leukocytes, and macromolecule leakage. The nature of these intercellular contacts varies substantially depending on the vessel size and location. For example, tight junctions are well developed in the blood—brain barrier but are less structurally defined in postcapillary venules, where fluid and solute transport is active. Capillaries and postcapillary venules respond to vasoactive agents, including vascular endothelial cell growth factor (VEGF), histamine, and prostaglandins, with increased flux through these sites.34 The tight junctions found in arteries tend to be more occlusive but may also be influenced by various agonists. Dynamic regulation of these pathways enables the endothelium to serve as a selective barrier, modulating access of highly mitogenic, thrombotic, or vasoactive substances to the underlying vascular smooth muscle.
Transendothelial vesicular transport is mainly used by the cell to transfer water-soluble macromolecules from the luminal surface to the abluminal surface. It has recently been shown that caveolae, vesicles containing the structural protein caveolin that are pinched off from the plasma membrane, are involved in transendothelial transport of macromolecules.35,36 Caveolae are also sites where a variety of kinases, docking proteins, G proteins, and receptors reside37 and therefore play an extremely important role in endothelial cell signal transduction.
Another major mechanism modulating endothelial barrier formation is endothelial cell contraction, analogous to smooth muscle contraction. This occurs in response to a variety of agonists, including thrombin, histamine, and ionomycin, and results in cell shape change that opens gap junctions between cells. It is likely that this contractile response is a major mechanism for edema formation in response to histamine and bradykinin and is involved in solute transport. This phenomenon is mediated by a series of intracellular signaling events, including activation of protein kinase C, myosin light chain phosphorylation, activation of tyrosine kinases, and stimulation of the small G protein Rho.38-40
Thus, the endothelium has both passive and active roles in the control of vascular permeability by acting as a physical permeability barrier and by modulating the expression of cell surface and secreted agonists and molecules that are capable of altering permeability.
The endothelium serves a dual function in the control of vascular tone (Fig. 8–2). It secretes relaxing factors such as NO, prostacyclin, and the endothelium-derived hyperpolarizing factor, as well as constricting factors such as endothelin. Vessel tone thus depends on the balance between these factors, as well as on the ability of the SMCs to respond to them. The most important regulatory molecules are discussed separately.
Figure 8–2.
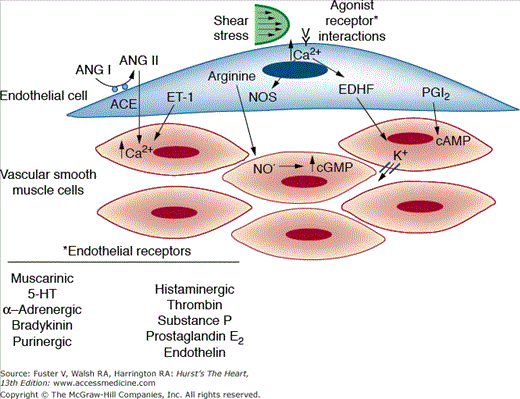
Endothelial control of vascular tone. Endothelial cells synthesize and secrete both vasodilator substances (nitric oxide [NO], endothelial-derived hyperpolarizing factor [EDHF], and prostaglandin I2 [PGI2]) and vasoconstrictor compounds (Ang II and ET-1). Secretion of these factors occurs in response to receptor stimulation and hemodynamic forces such as shear stress. Vessel tone depends on the balance between these factors, as well as on the ability of the smooth muscle cells to respond to them. ACE, angiotensin-converting enzyme; ANG, angiotensin; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; ET-1, endothelin-1; 5-HT, 5-hydroxytryptamine; NOS, nitric oxide synthase.
An endothelium-derived relaxing factor (EDRF) was first described by Furchgott and Zawadzki,2 who observed that aortic rings dilated in response to acetylcholine only when the rings maintained an intact endothelium. The EDRF was subsequently found to be NO.41
NO is produced by the action of the enzyme nitric oxide synthase (NOS), which oxidizes the guanidino nitrogens of L-arginine to form citrulline and NO. This enzyme has been cloned from brain (nNOS, for neuronal NOS, type I),42 macrophages (iNOS, for inducible NOS, type II),43 and endothelial cells (eNOS, for endothelial NOS type III).44 The three isoforms of NOS share important consensus sequences for nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide, and flavin mononucleotide cofactor-binding sites, as well as a Ca2+-calmodulin binding site. An important cofactor for the NO synthases is tetrahydrobiopterin, which participates in electron transfer from the heme group of the enzyme to L-arginine. Interestingly, when tetrahydrobiopterin or L-arginine is absent, electron transfer is shunted to molecular oxygen, resulting in formation of the superoxide anion.45 This phenomenon has been termed uncoupling of NOS, and substantial data suggest that this may occur in a variety of disease states.46
Many factors have been shown to regulate the release of NO.47 These include hormones such as acetylcholine, norepinephrine, bradykinin, thrombin, adenosine triphosphate (ATP), and vasopressin; the platelet-derived factors serotonin and histamine; fatty acids; ionophores; and shear stress. NO easily crosses the SMC membrane and binds to the heme moiety of the soluble guanylate cyclase, thereby enhancing the formation of cyclic guanosine monophosphate (GMP). Cyclic GMP, in turn, reduces intracellular Ca2+ concentrations, leading to dephosphorylation of the myosin light chain and relaxation.48 It should be noted that the drug nitroglycerin exerts its vasodilator effects by being converted to NO, thus substituting for a natural product.
Although increases in intracellular calcium in response to the aforementioned agents clearly activate eNOS via binding of Ca and calmodulin, phosphorylation of the enzyme is important in regulating its activity. For example, shear stress acutely stimulates the release of NO from the endothelium, and this depends only on calcium during the first few seconds of the response.49 The continued activation of eNOS in response to several minutes or hours of shear is maintained by serine phosphorylation.50
The expression of eNOS is highly regulated. Increases in shear stress enhance eNOS expression, and low levels of shear decrease it.51 Exercise training dramatically increases eNOS expression in endothelial cells, likely because of the increased shear stress caused by the high cardiac output that accompanies sustained exercise.52 In contrast, oxidized LDL, hypoxia, and inflammatory cytokines such as tumor necrosis factor-α (TNF-α) decrease eNOS expression.44,53,54 The hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors increase eNOS levels by stabilizing the eNOS mRNA. This is thought to be an important component of the so-called pleiotropic effects of the statins that may contribute to their therapeutic effects.
Shortly after the identification of NO, it was suspected that the endothelium could release more than one relaxing factor, depending on the vessel size, stimulus, and species studied. Initial studies showed that some vasodilators produce hyperpolarization of the vascular smooth muscle membrane in an endothelium-dependent manner. It is now clear that this is caused by the release of hyperpolarizing factors from the endothelium that are different from NO.55 Increasing evidence suggests that there are at least two EDHFs. One is 14,15-epoxyeicosatrienoic acid (14,15-EET), a cytochrome P450 metabolite of arachidonic acid.56 This epoxide and its metabolite 14,15-dihydroxyeicosatrienoic acid are released from the endothelium, diffuse to the adjacent vascular smooth muscle, and open calcium-activated potassium channels. This results in efflux of potassium ions, hyperpolarization, and subsequent closing of voltage-dependent calcium channels, leading to vasodilatation.57 The second EDHF is likely hydrogen peroxide, which is also made by endothelial cells and acts in the same fashion as the EETs.58,59 Recent studies have shown that hydrogen peroxide is particularly important in human coronary arterioles.60 In general, as one progresses from large conduit vessels to smaller arterioles, the role of NO seems to decrease, and the EDHF component of vasodilatation increases.61 Moreover, there is an interplay between regulation of NO levels and EDHF production. NO can bind to the heme group of the cytochrome P450s and inhibit these enzymes and their production of 12,15-EET. If NO production is diminished, this seems to unleash cytochrome P450 EET production, which can then take over the role of NO in mediating vasodilatation. In the setting of excessive superoxide production, NO is oxidatively degraded, leading to loss of NO bioactivity. A substantial portion of the increased superoxide is dismutated to hydrogen peroxide, which can then also serve as an EDHF. Through these mechanisms, the loss of NO can lead to a compensatory increase in EDHF and maintenance of small vessel vasodilatation.
Prostacyclin, or prostaglandin I2 (PGI2), a prostanoid derived from the action of cyclooxygenase (COX)-1 and -2 on arachidonic acid, is released by the endothelium and relaxes vascular smooth muscle by increasing its intracellular content of cyclic adenosine monophosphate.62 Prostacyclin is also platelet suppressant and antithrombotic and reduces the release of growth factors from endothelial cells and macrophages.25 Inhibition of these beneficial effects likely underlies the untoward clinical outcomes observed in patients treated with COX-2 inhibitors.63 Among the agonists that stimulate prostacyclin synthesis are bradykinin (one of the most potent), substance P, platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and adenine nucleotides.25 Prostacyclin has been shown to compensate for the loss of NO in the eNOS knockout mouse.64 Analogues of prostacyclin such as iloprost have proven useful in the treatment of pulmonary hypertension.65
Although the endothelium predominantly produces prostacyclin, under pathologic conditions, it can begin to generate other prostaglandins with vasoconstrictor activity.66 These include prostaglandin H2 (PGH2), a direct product of COX-1 and -2, and thromboxane, made by the action of thromboxane synthase on PGH2.67 In hypertensive models, various hormones such as acetylcholine and endothelin-1 can stimulate the release of an endothelium-derived constricting factor (EDCF) that acts on the thromboxane receptor. Interestingly, EDCF is not thromboxane but the endoperoxide PGH2, and paradoxically, in some models may be prostacyclin, which causes constriction because of a loss of the PGI2 receptor.68
Another important vasoconstrictor derived from cytochrome P450 metabolism of arachidonic acid is 20-hydroxyeicosatetraenoic acid (20-HETE).69 This compound depolarizes vascular SMCs by inhibiting calcium-activated potassium channels and thereby promotes vasoconstriction. The synthesis of 20-HETE is stimulated by angiotensin II, endothelin, and catecholamines and is inhibited by NO.69 Not surprisingly, 20-HETE production is elevated in a number of common diseases such as hypertension, kidney disease, and diabetes.
Endothelial cells, particularly those in the pulmonary vasculature, synthesize and express angiotensin-converting enzyme (ACE) on their surface.70 ACE converts angiotensin I to the potent vasoconstrictor angiotensin II and degrades and inactivates bradykinin. Recent work has suggested that upon binding of ACE inhibitors, ACE can directly signal via its short cytoplasmic tail, leading to changes in gene expression.71 The overall importance of this phenomenon in the cardiovascular system is unclear. Of note, vascular and cardiac cells contain almost all components of the renin/angiotensin system,72 and thus local production of angiotensin II can contribute importantly to vascular function. This local production of angiotensin II may explain why ACE inhibitors and angiotensin receptor antagonists are often effective even when the circulating levels of renin or angiotensin II are not elevated.
Recently, a new carboxypeptidase, ACE-II, has been identified.73 This enzyme cleaves one amino acid from either angiotensin I or angiotensin II. The overall effect of ACE-II action is a reduction in angiotensin II and an increase in the metabolite angiotensin I-VII, which has vasodilator properties. Thus, the balance between ACE and ACE-II is an important factor controlling angiotensin II levels and ultimately vasomotor tone.74
The endothelins are a family of closely related peptides made and secreted by many cells, including endothelial cells. There are three endothelins (ET-1, -2, and -3), all of which are 18 amino acid peptides. The endothelins are initially synthesized as preproendothelin, which undergoes preprocessing to big endothelin. Big endothelin is released and converted to active endothelin by the endothelin-converting enzyme. The synthesis of preproendothelin is stimulated by diverse stimuli, including angiotensin II, oxidized LDL, hypoxia, low shear stress, and inflammatory cytokines.75 The vascular effects of endothelin are mediated by endothelin receptors, of which three subtypes have been identified: A, B, and C. The receptors have differing specificity for the individual endothelin peptides and activate different signaling pathways. In the vessel, the ET-A receptor exists predominantly on vascular smooth muscle, and the ET-B receptor resides on endothelial cells. Activation of the former stimulates potent vasoconstriction, and activation of the latter stimulates release of NO and thus favors vasodilation.76
ET-1 causes a slow, intense, and sustained contraction likely resulting from activation of the phosphoinositide/protein kinase C signaling pathway and opening of the voltage-dependent L-type calcium channels.77 Importantly, even low, subthreshold concentrations of ET-1 enhance vasoconstriction by other agents, including serotonin, angiotensin II, and α-adrenergic agonists, seemingly via activation of protein kinase C.
ET-1 is also a potent growth factor for SMCs78 and a chemoattractant for monocytes.79 Angiotensin II has been shown to stimulate the production of ET-1 by SMCs in culture,80 and in vivo some of the hypertensive effect of angiotensin II is mediated by endothelin.81 Thus, ET-1 and angiotensin II act in concert in conditions such as hypertension, diabetes, and heart failure.82,83 Currently, the mixed ET-A and ET-B receptor antagonist bosentan is clinically used for patients with pulmonary hypertension.75
Many of the previously described endothelial functions are modulated by the physical forces of stretch, strain, and shear stress imposed by the hemodynamics of the circulation. Both stretch of the vessel wall (as observed in hypertension) and shear stress have been shown independently to affect endothelial cell morphology, function, or both. Studies in cultured cells have shown that stretching endothelial cells leads to changes in cell shape,84 intracellular signal generation with an increase in calcium and superoxide levels,85 and proliferation.86 Shear stress has numerous effects on endothelial cells. Initially, it was found that exposure of endothelial cell monolayers to elevated shear stresses in vitro caused them to align in the direction of flow. This reorientation was accompanied by changes in the cytoskeleton of the cells, including reorganization and alignment of the actin filaments and microtubules (Fig. 8–3). Similar mechanisms presumably also account for the orientation of endothelial cells parallel to the longitudinal axis in areas of laminar flow in the arterial system. The function of the endothelium is also altered by shear stress. Some of the cellular responses to shear stress include activation of K+ currents; increased secretion of vasoactive and growth factors, including NO, endothelin, prostacyclin, and basic fibroblast growth factor (bFGF); enhanced tissue factor expression; elevation of LDL uptake; and increased tPA secretion.87
Figure 8–3.
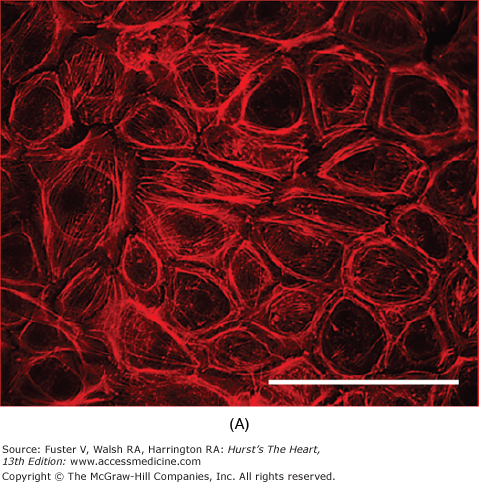

Effect of shear stress on endothelial cells. In bovine aortic endothelial cells grown in static conditions, F-actin filaments assume a random orientation as visualized by rhodamine-labeled phalloidin staining (A). On exposure to shear stress (30 dynes/cm2, 24 h), these filaments align (B). Bars, 100 μm. Courtesy of Lula Hilenski, PhD.
The importance of these observations lies in the variation in hemodynamic forces throughout the circulation. Areas of the vasculature exposed to low shear stress (branch points and curvatures) exhibit a predilection to the formation of atherosclerotic lesions.88 True oscillations of flow have also been shown to occur in the carotid bulb, proximal coronary arteries, and distal aorta.89 Studies in cultured endothelial cells have shown that oscillatory shear stress increases endothelial cell production of reactive oxygen species (ROS),90 enhances adhesion molecule expression, and stimulates monocyte adhesion.91 Recent work has shown that the bone morphogenic protein 4 is a key signaling molecule in the pathways leading to these events.92,93
The mechanisms by which the endothelial cell can sense and transduce mechanical signals have been extensively studied. Two recent hypotheses have emerged suggesting that mechanical forces are transduced by extracellular glycosaminoglycans or by the selectin, platelet endothelial cell adhesion molecule-1 (PECAM-1). Enzymatic disruption of the glycocalyx has been shown to decrease endothelial cell NO production in response to shear and to alter cell motility.94,95 Coexpression of PECAM-1 with VE-cadherin confers shear responsiveness in heterologous cells that normally are not affected by shear stress.96 These mechanosensors in turn activate integrins that modify the cytoskeleton and coordinate downstream signaling. As an example, integrin activation leads to stimulation of kinases and phosphatases in focal adhesion complexes.97 Changes in the actin cytoskeleton may affect RNA stability and translation.98 In addition, flow-sensitive ion channels99 and G proteins participate in mechanotransduction.100 Furthermore, caveolae, which are flask-shaped membrane vesicular structures, are rich in signaling molecules such as G proteins, and are involved in signal generation in response to shear stress.101
Physiology of the Vascular Smooth Muscle Cell
SMCs normally respond to hormonal stimulation with contraction or relaxation. In certain disease states, however, growth or hypertrophy and migration to the intima are predominant responses. Some of the biochemical signals generated by these vasoactive agonists are similar for both types of responses, with the final physiologic response dictated by the phenotype and environment of the cell, and the exact biochemical pathways activated.
Some of the earliest signals generated within the cell after stimulation with vasoactive agonists involve hydrolysis of a specific class of membrane lipids, the phosphoinositides, by phospholipase C.102 This event leads to production of inositol triphosphate (IP3) and diacylglycerol (Fig. 8–4). IP3 binds to IP3 receptors (IP3Rs) on intracellular organelles with releasable calcium stores. When IP3 binds to these receptors, they form channels, allowing calcium transit from these organelles into the cytoplasm, where it stimulates molecular pathways, leading to contraction.103 IP3R activity is modulated not only by IP3 binding but also by phosphorylation and binding of nucleotides such as ATP and NADH.104 Intracellular calcium levels can oscillate and are increased by activation of calcium influx via L-type CaV1.2 channels or transient receptor potential (TRP), an event necessary to maintain calcium stores.105 The consequent increase in cytosolic Ca2+ activates a cascade of enzymes leading to contraction or growth (see below). Calcium signaling is terminated by reuptake of calcium into the endoplasmic reticulum via the sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) and by extrusion of Ca2+ via the plasma membrane Ca2+-ATPase.105
Figure 8–4.
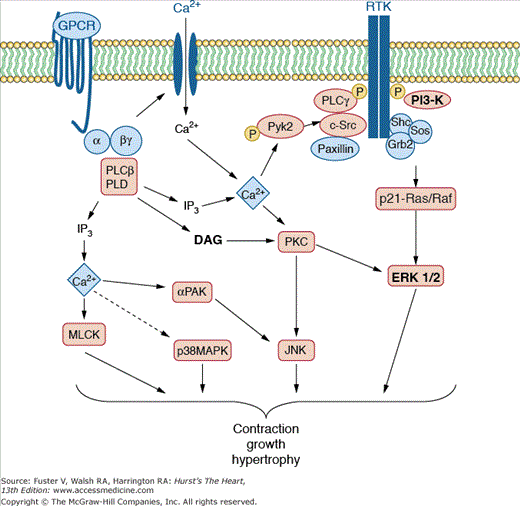
Signaling pathways in vascular smooth muscle. Vasoconstrictor agonists interact with specific G protein—coupled receptors (GPCRs) on vascular smooth muscle. These receptors are linked to a heterotrimeric G protein (αβγ), which then couples to one or more phospholipase Cs (PLCs) or phospholipase D (PLD). PLC cleaves the inositol phospholipids to yield diacylglycerol (DAG) and inositol phosphates, particularly inositol triphosphate (IP3). IP3 releases calcium from intracellular stores, and along with DAG, activates the Ca2+ and phospholipid-dependent enzyme protein kinase C (PKC). Ca2+ activates numerous other kinases, including p21-activated kinase (α-PAK), Pyk2, and myosin light-chain kinase (MLCK). PLD cleaves phosphatidylcholine to release phosphatidic acid, which is converted to DAG. PKC is involved in activation of the mitogen-activated protein kinase (MAPK) cascade, including extracellular signal-regulated kinases (ERK 1/2) and Jun kinase (JNK). Growth factors activate receptor tyrosine kinases (RTKs), Src, PLC-γ, and phosphatidylinositol 3-kinase (PI3-K). RTKs also phosphorylate and form a signaling complex with paxillin and adapter proteins such as Shc, which binds Grb-2 and Sos and ultimately mediates the conversion of Ras to its active form, Ras phosphorylates Raf1, which in turn leads to activation of the MAP kinase cascade. Courtesy of Bernard Lassègue, PhD.
Diacylglycerol, the other product of phospholipase C activation, is a potent activator of protein kinase C, a Ca2+– and phospholipid-dependent enzyme that phosphorylates numerous cellular proteins and thereby enhances contraction at any given level of intracellular calcium.106 Diacylglycerol can be further metabolized to phosphatidic acid or to glycerol, fatty acids, and ultimately eicosanoids and leukotrienes that may themselves modulate tone.
Contractions induced by various vasoactive hormones differ not only in magnitude and time course but also differ between vessels. In general, there is an initial, rapid component of force generation and a more sustained phase of contraction. Some agonists, such as angiotensin II, induce only a transient constriction of many vessels, but others, including norepinephrine, endothelin, and vasopressin, cause sustained contractions. The initial phase of force development depends on formation of actin—myosin cross-bridges in response to acute elevations of intracellular calcium, and the sustained phase of contraction persists even after calcium levels return toward baseline.
A sliding-filament mechanism similar to that found in skeletal muscle is thought to regulate phasic contraction of smooth muscle. Tension development is regulated by myosin light-chain kinase (MLCK)—mediated phosphorylation of myosin light chain (Fig. 8–5). When Ca2+ increases within the cell in response to hormonal stimulation, it binds to calmodulin, which in turn associates with MLCK, converting it from an inactive to an active form. MLCK then phosphorylates the MLC, enabling actin to activate myosin Mg2+-ATPase and crossbridge formation. When the intracellular Ca2+ concentration drops to less than approximately 100 nM, Ca2+ dissociates from calmodulin, calmodulin detaches from MLCK, and MLCK becomes inactive. MLC phosphatase activity then predominates, myosin is dephosphorylated, and crossbridge cycling ceases. During sustained contraction, however, the intracellular Ca2+ concentration is low, and energy consumption is reduced, suggesting the development of a latch-bridge or of a low cycling state.107 Alternatively, the Ca2+ sensitivity of the contractile apparatus may be increased, a response posited to be regulated by protein kinase C.108 This latch state is also modulated by the actin-binding proteins caldesmon and calponin.109 Caldesmon tonically inhibits contraction. Agonists such as phenylephrine stimulate extracellular regulated kinase (ERK) 1/2—mediated phosphorylation of caldesmon and enhance binding of calcium/calmodulin, removing its inhibitory effect and increasing tension.110 Calponin has been suggested to directly inhibit the ATPase activity of myosin and to act as a signaling molecule that facilitates agonist-stimulated activation of protein kinase C.111,112
Figure 8–5.
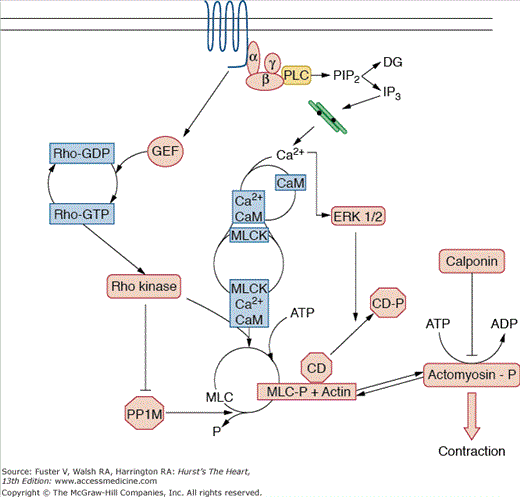
Contraction cascade. Activation of smooth muscle by a vasoconstrictor hormone leads to a cascade of biochemical signals, ultimately resulting in phosphorylation of actomyosin, crossbridge formation, and force generation. The release of Ca2+ from intracellular stores is one of the major initiating events because Ca2+ combines with calmodulin (CaM) to activate myosin light-chain kinase (MLCK). This enzyme phosphorylates the myosin light chain (MLC), which is then able to interact with actin. In addition, activation of a guanine nucleotide exchange factor (GEF) for the small—molecular-weight G protein Rho leads to stimulation of Rho and Rho kinase, which inhibits myosin phosphatase (PP1M), thus enhancing MLC phosphorylation (MLCP). Caldesmon (CD), which normally inhibits actin—myosin interaction, becomes phosphorylated by extracellular signal regulated kinase (ERK 1/2) and is released from this complex. Calponin acts by inhibiting myosin ATPase activity. αβγ, heterotrimeric G protein; ATP, adenosine triphosphate; Ca2+, calcium; DAG, diacylglycerol; IP3, inositol triphosphate; P, phosphate; PIP2, phosphatidylinositol 4,5-bisphosphate; PLC, phospholipase C. Courtesy of Bernard Lassègue, PhD.
In addition to this classical regulation of SMC contraction, the small—molecular-weight guanosine triphosphatase (GTPase) Rho, initially described as a modulator of the actin cytoskeleton, is an important regulator of contractility. In its active GTP-bound form, Rho activates Rho kinase, which inhibits myosin phosphatase type 1.113 This inhibition in turn sustains MLC phosphorylation and sensitizes the contractile apparatus to calcium.114 Because its activity seems to be increased in hypertension,115 Rho kinase has become a target of therapeutic interventions. Inhibitors of Rho kinase have been shown to lower vascular resistance and thus blood pressure in patients with hypertension.116,117
Counteracting the signaling pathways promoting contraction are intracellular cyclic nucleotides, such as cyclic AMP and cyclic GMP. As noted above, NO binds to soluble guanylate cyclase in SMCs, decreases intracellular calcium, and therefore reduces myosin light chain phosphorylation. Degradation of cyclic nucleotides is catalyzed by 3′,5′-cyclic nucleotide phosphodiesterases (PDEs), particularly PDE3 and PDE5 in SMCs.118 PDE5 inhibitors, such as the impotence drug sildenafil, enhance cyclic GMP concentrations and have mild hypotensive effects. They have been approved for the treatment of pulmonary hypertension.119
Normally, vascular SMCs are relatively refractory to growth stimuli and exist in a quiescent, differentiated state. The healthy endothelium is critically important in maintaining this phenotype. Products of the endothelium, such as NO,120 prostacyclin,120 heparan sulfates,121 and transforming growth factor-β (TGF-β),122 directly inhibit vascular smooth muscle growth, and some, such as TGF-β, actively promote differentiation. One of the most important intracellular signaling pathways that keeps SMCs in the differentiated state is cyclic GMP-dependent protein kinase (PKG). PKG expression promotes the differentiated phenotype, and loss of PKG is a characteristic of proliferating SMCs.123 The endothelium is also an effective barrier limiting access of bloodborne growth factors to vascular smooth muscle. For example, the antithrombotic properties of the endothelium prevent access of promitogenic factors such as PDGF and thrombin to the underlying smooth muscle. Endothelial disruption allows initiation of a mitogenic smooth muscle response and regrowth of normal endothelium inhibits further proliferation.124 In addition to these effects of the normal endothelium, the healthy vascular matrix minimizes vascular smooth muscle proliferation.
Under pathophysiologic conditions, the vascular milieu begins to favor vascular smooth muscle growth. One important phenomenon is degradation of the ECM to allow SMC migration, proliferation, and hypertrophy.125 This is largely caused by MMPs released by activated cells intrinsic to the vessel or as invading inflammatory cells. A second pathologic alteration is the secretion of promitogenic agents by cells intrinsic to the vessel and invading inflammatory cells. The best studied of these factors is PDGF, so named because it was originally isolated from platelets. PDGF is composed of two distinct peptide chains (designated A and B chains) and can be secreted as an AB heterodimer or as an AA or BB homodimer. Release of PDGF from the endothelium is regulated by growth factors, including TGF-β, fibroblast growth factor (FGF), and TNF; circulating factors; and locally produced factors such as thrombin.126 Another growth factor is insulin-like growth factor 1 (IGF-1),13 which is a progression factor that facilitates movement of cells through the cell cycle and enhances the mitogenic effect of PDGF on smooth muscle.127 IGF-1 production by endothelium is regulated by PDGF and plays a major role in vascular hypertrophy and hyperplasia.128 Other factors that affect smooth muscle proliferation include interleukin-1 (IL-1), FGF, and endothelin. IL-1 is an inflammatory cytokine that has numerous vascular effects in addition to mitogenesis, including the stimulation of procoagulant activity,129 induction of leukocyte adhesiveness (see below), and inhibition of contraction.130 Basic FGF acts as a potent smooth muscle mitogen, particularly after denuding injury.131 It is stored in the subendothelial matrix and may be released by heparin and proteinases,132 suggesting that the matrix may serve as a store for rapidly mobilizing this growth factor. FGF released from SMCs may be particularly important in the growth response induced by injury to the arterial wall. Finally, endothelin-1, through its action on the ET-A receptor, induces SMC growth by stimulating increases in intracellular calcium, activating protein kinase C, and increasing intracellular production of ROS. Diverse stimuli such as elevated insulin,133 oscillatory shear stress and pressure,134 and angiotensin II135 potently induce endothelial production of ET-1.
Vascular SMC growth occurs via two processes: hypertrophy and hyperplasia. In general, whereas hypertrophy occurs in response to long-term stimulation with vasoconstrictor-type agents, hyperplasia occurs in response to the classical growth factors. Hypertrophy is characterized by an increase in SMC mass caused by increased protein synthesis without an accompanying replication of DNA and has been shown to occur in response to angiotensin II136 and thrombin137 as well as in large vessels during hypertension. Hyperplasia is characterized by cell replication and is stimulated by growth factors such as PDGF and FGF after vascular injury.131,138,139 The biochemical processes leading to hypertrophy and hyperplasia share some similarities but are not identical.
Classic growth factors, such as PDGF, promote growth both by suppressing the contractile phenotype140 and by activating many of the same signaling pathways as do vasoconstrictors: phosphoinositide hydrolysis, Ca2+ mobilization and influx, and protein kinase C activation. Receptors for these growth factors are intrinsic tyrosine kinases, leading to the tyrosine phosphorylation of numerous proteins that are essential for growth. Tyrosine phosphatases can counteract the mitogenic effects of growth factors by inhibiting tyrosine phosphorylation of specific substrates.141
When growth factor receptors are activated, they recruit a complex of proteins that subsequently activate multiple signaling cascades, leading to the final cellular response.142 Initially, growth factor receptors dimerize and phosphorylate themselves on tyrosine residues. Some proteins, such as phospholipase C-γ, the tyrosine kinase c-Src, and phosphatidylinositol 3-kinase, bind directly to receptor tyrosine kinases, but others, including the tyrosine kinase Pyk-2 and the cytoskeletal protein paxillin, associate with the receptor via linker proteins such as Grb and Shc. Shc and Grb2 link these receptors to Ras, a ubiquitous GTPase that initiates a serine/threonine kinase cascade that includes mitogen-activated protein kinases (MAPKs) and ultimately leads to growth. Another important kinase is c-Abl, an Src substrate that activates Rac and therefore cytoskeletal rearrangement and gene expression.143 Many of these proteins are also activated by seven transmembrane-spanning G protein—coupled receptors,144,145 an observation that may partially explain the growth-promoting properties of vasoconstrictor hormones such as angiotensin II, thrombin, and ET-1. Although full growth factors such as PDGF ultimately promote cell cycle progression and proliferation by activating cell cycle proteins (cyclins and cyclin-dependent kinases) and inhibiting cell cycle inhibitors (p27kip1), hypertrophic agents such as angiotensin II have a much more modest effect on cyclins and increase the expression of p27kip1.146
ROS play a crucial role in modulation of these growth-related signaling pathways. Growth-promoting agonists stimulate the NADPH oxidases Nox1 and Nox5 to produce superoxide and hydrogen peroxide,147,148 which may serve as progenitors to numerous other ROS.149,150 Of these, hydrogen peroxide seems to be particularly important in the growth process. Production of small concentrations of endogenous hydrogen peroxide activates specific mitogenic signaling molecules such as Src, Ras, p38MAPK, and Akt/protein kinase B via oxidation or glutathiolation and promotes entry into the cell cycle.151-155 More importantly, hydrogen peroxide inactivates protein tyrosine phosphatases by oxidizing critical cysteine residues, leading to sustained protein phosphorylation of target molecules. Initial inactivation of phosphatases such as phosphatase and tensin homolog (PTEN) by hydrogen peroxide enhances phosphorylation of and signaling through phosphatidylinositol 3-kinase.156 This inhibition of phosphatase activity is limited by peroxiredoxins, important scavengers of hydrogen peroxide that are localized in specific regions of the cell. One of these, peroxiredoxin II, has a unique role in modulating cell growth because it binds to the PDGF receptor and reduces local hydrogen peroxide concentrations, thereby releasing phosphatase inhibition and terminating signaling.157
The Extracellular Matrix
The ECM is a major component of the vessel wall. It is the medium through which nutrients are transported; a repository for products secreted by the cells of the vascular wall; the site of accumulation of cell debris; and a substrate for migration and proliferation of endothelial cells, monocytes, and vascular SMCs. The matrix consists of multiple proteins that have distinct functions in maintaining the integrity and function of the vessel wall (Table 8–1). Interactions between the multiple constituents of the ECM and the vascular cells generate complex, highly organized signals mediated by specific membrane receptors. These signaling networks affect outcomes as diverse as organ development, dynamic remodeling such as hypertrophy and angiogenesis, and inflammatory diseases such atherosclerosis. The developmental aspects of extracellular matrix—cell interactions were reviewed recently.158



