(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Several important mechanisms are involved in the mitochondrial response to varying cellular energy demands. In cardiomyocyte, a short-term adjustment can be made through cytosolic processes including metabolic signaling and signal transmission. The former one is achieved by changing cytosolic ADP and Pi concentrations (ATP hydrolysis in the cytosol increases ADP and Pi fluxes to mitochondria, and the amount of substrates available for ATP production increases). Metabolic signaling adjustments are made via phosphotransfer networks and compartmentalized energy transfer systems. The mitochondrial adenine nucleotide translocator plays a central role in the ATP transfer from mitochondria to the cytosol. Impairment of this essential transport system is implicated in several neuromuscular diseases. In addition, direct functional interplay exists in cardiomyocytes between mitochondria and other subcellular compartments that makes it possible direct energy and signal channeling between organelles.
Ca2+ is a signal transmitter linking mitochondrial energy production to cellular energy utilization. ATP hydrolysis is coupled to changes in free cytosolic Ca2+ and mitochondrial Ca2+. Consistent with this notion is the localization of mitochondria near Ca2+ release sites. The latter activate mitochondrial enzymes taking part in ATP production (e.g., several dehydrogenases, ATP synthase).
In this chapter, the mechanisms that coordinate the activation of oxidative phosphorylation by many signaling molecules are discussed in the context of recent discoveries of various regulatory posttranslational modifications of mitochondrial proteins.
Introduction
In cardiac cells, specialized cellular functions are highly organized within structural and functional compartments. Major energy-consuming processes are localized to the sarcoplasmic reticulum and myofibrillar compartments, while energy production takes place mainly within mitochondria. Several mechanisms ensure efficient energy transfer and communication between sites of energy production and energy utilization. Moreover, a number of complex regulatory mechanisms have been evolved to synchronize mitochondrial energy production with cellular energy demand.
It has been estimated that mitochondrial functioning requires the biosynthesis of approximately 1,500 polypeptides, most of which are encoded by nuclear DNA (nDNA). Promoters of many nuclear genes, encoding components of oxidative phosphorylation (OXPHOS) and enzymes involved in processing high-energy lipids, contain binding sites for transcription factors regulated by various external signals (availability of nutrients, energy deficit). Thus, regulation of nuclear gene expression of mitochondrial proteins tightly controls mitochondrial energy production in accordance with constantly changing cell requirements. For instance, upregulation of the peroxisome proliferator-activated receptor α (PPARα) upregulates mitochondrial fatty acid (FA) β-oxidation (FAO) and concomitantly downregulates glucose uptake and oxidation.
Energy supplies and several other signaling molecules are very important regulators of mitochondrial functioning. In the heart, substrate availability can significantly alter mitochondrial metabolism: increases in circulating free FAs have been shown to decrease glucose oxidation. The control of mitochondrial energy conversion by cytosolic processes includes cytosolic signaling molecules ADP, Pi, and Ca2+. The exchange of extramitochondrial ADP and intramitochondrial ATP across the mitochondrial inner membrane (MIM) is facilitated by adenine nucleotide translocator (ANT). Thus, ANT plays an important role in the regulation of the intracellular energetic balance, and dysfunction of ANT is implicated in neuromuscular diseases. On the other hand, ATP channeling is the way for mitochondria to interact with other organelles to influence their functions. Growing evidence suggests the existence of communications between mitochondria and the SR, between mitochondria and myofilaments, and between mitochondria and nucleus. Indeed, mitochondria appear to be clustered at sites of high ATP demand and are organized into highly ordered elongated bundles, wrapped around the myofibrils and in contact with the SR and nucleus. Direct mitochondrial ATP supply and/or ADP withdrawal is nearly as effective as creatine kinase-supplied ATP and is much more effective than externally supplied ATP, in supporting calcium uptake and contractile velocity of cardiomyocytes.
Ca2+ is one of the major signaling molecules acting both as a feedforward and feedback indicator of cellular ATP hydrolysis. It also acts on numerous targets within mitochondria to maintain tightly balanced ATP production. Furthermore, extracellular regulation of cardiac mitochondrial functioning is achieved via hormones/posttranslational modifications.
In this chapter, we will discuss the mechanisms of the coordinated activation of OXPHOS by extracellular signaling molecules in the light of the recent discoveries of various regulatory posttranslational modifications of mitochondrial proteins.
Mitochondria/Nucleus Interactions
The nuclear genome in coordination with the mitochondrial genome regulates mitochondrial function, dynamics, and number necessary to maintain oxidative metabolism and ROS generation, utilization, and breakdown. This regulation occurs via a number of nuclear regulatory proteins that control transcription of components of mitochondrial multi-subunit respiratory complexes, ATP synthase, etc. Nuclear regulatory proteins include nuclear respiratory factors (NRFs) 1 and 2, the cyclic AMP response element-binding protein (CREB), and transcription factor A of mitochondria (TFAM) [1, 2]. Nuclear regulatory protein-dependent activation of target genes encoding subunits of OXPHOS is modulated by upstream transcription factors, including the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1 α and β (PGC1α and PGC1β, respectively) and PGC1-related coactivator (PRC) [3, 4]. Overexpression of PGC1α, for instance, causes uncontrolled increase in mitochondrial number leading to impairment of cardiomyocyte contractile function [5]. Conversely, knockout of PGC1α results in diminished ATP production and inability of the myocardium to respond to exercise or to β-adrenergic stimulation [6]. Expression of PGC-related factors is regulated by many external signals, like availability of nutrients, energy deficit, and temperature (Table 6.1) [7, 8].
Table 6.1
Nuclear factors implicated in the expression of mitochondrial bioenergetics machinery
DNA-binding transcription factors | ||||
Target genes | References | |||
Nuclear respiratory factor (NRF)-1 | Complex I | NDUFS8 (human) | ||
Complex II | SDH-A | [10] | ||
SDH-B | ||||
SDH-C | ||||
SDH-D | ||||
Complex III | Ubiquinone-binding protein (human) | |||
Core protein I (human) | ||||
Complex IV | Subunit Vb (mouse, rat, human/primate) | |||
Subunit VI-H (rat) | ||||
Subunit VIa-L (human) | ||||
Subunit VIIa-L (bovine) | ||||
Cytochrome c | (Rat, human) | |||
ATP synthase | γ Subunit (bovine) | |||
c Subunit (human) | ||||
Nuclear respiratory factor (NRF)-2 | Complex II | SDH-B (human) | ||
SDH-C (human) | ||||
SDH-D (human) | ||||
Complex IV | Subunit IV (rat, mouse) | |||
Subunit Vb (mouse, rat, human/primate) | ||||
Subunit VIa-L (human) | ||||
Subunit VII-H (bovine) | ||||
Subunit VIIa-L (bovine, human) | ||||
ATP synthase | β Subunit (human) | [12] | ||
Mitochondrial glycerol phosphate dehydrogenase | (Human) | [13] | ||
Sp1 | Complex II | SDH-A | [10] | |
SDH-B | [10] | |||
SDH-D | [10] | |||
Complex IV | Subunit IV | [12] | ||
Subunit Vb | [14] | |||
Subunit VIIa-H (human) | [15] | |||
Subunit VIIa-L (bovine) | [16] | |||
Cytochrome c | (Rat) | [17] | ||
ATP synthase | α Subunit | [18] | ||
β Subunit | [19] | |||
PDHC | PDHK4 | [20] | ||
α-ketoglutarate dehydrogenase | Dihydrolipoyl succinyltransferase (E2) | [21] | ||
ATF/CREB | Complex IV | Subunit IV (rat) | [22] | |
Cytochrome c | (Rat) | |||
α-ketoglutarate dehydrogenase | Dihydrolipoyl succinyltransferase (E2) | [21] | ||
Dihydrolipoyl dehydrogenase (E3) | [23] | |||
Yin Yang 1 (YY1) | Complex IV | Subunit Vb (murine) | ||
Subunit VIa-L (human) | [25] | |||
Subunit VII-H (bovine) | [26] | |||
ATP synthase | α Subunit | [27] | ||
OXBOX/REBOX | ATP synthase | ATP synthase β subunit | [28] | |
Myocyte enhancer factor-2 (MEF2) | Complex IV | Subunit VIa-H | ||
Subunit VIIa-H (human) | [15] | |||
MyoD | Complex IV | Subunit VIa-H | [30] | |
Subunit VIIa-H (human) | [15] | |||
Subunit VIII-H (rat) | ||||
Retinoic acid-bound nuclear retinoid X receptor (RXR)/retinoic acid receptor (RAR) | PDHC | PDHK4 | [20] | |
FOXO | PDHC | PDHK4 | [20] | |
Peroxisome proliferator-activated receptor (PPAR) α | PDHC | PDHK4 | ||
FA utilization enzymes | VLCAD | [34] | ||
LCAD | [34] | |||
MCAD | ||||
3-Hydroxyacyl-CoA dehydrogenase | [34] | |||
Glucocorticoid receptor (GR) | PDHC | PDHK4 | [20] | |
α-ketoglutarate dehydrogenase | Dihydrolipoyl succinyltransferase (E2) | [21] | ||
Estrogen-related receptor α (ERRα) | ATP synthase | β Subunit | [37] | |
FA utilization enzymes | MCAD | [4] | ||
Nuclear coactivators | Interaction with transcription factors | Interaction with other coactivators | Target genes | References |
Peroxisome proliferator-activated receptor gamma coactivator 1α (PGC1α) | NRF-1, PPARα, CREB, ERRα | SRC-1, CBP/p300, HCF | Cytochrome c oxidase subunit IV, cytochrome c, FA utilization enzymes | |
Peroxisome proliferator-activated receptor gamma coactivator 1β (PGC1β) | NRF-1, PPARα, hepatocyte nuclear factor 4α (HNF-4α) | HCF | Same | [38] |
PGC1-related coactivator (PRC) | NRF-1, CREB | HCF | Cytochrome c | |
Host cell factor (HCF) | NRF-2 | PGC1α, PGC1β, PRC | Cytochrome c oxidase subunit IV, ATP synthase β subunit | |
Seven subunits of complex I, one subunit of complex III, and three subunits of complex IV (CcO) are encoded by mtDNA. The majority of ETC proteins are encoded by nuclear genome. On nDNA genes, encoding PSST subunit of complex I, cytochrome c, and a number of subunits of complexes II, III, and IV, NRF-1-recognition sites have been demonstrated. Similarly, genes, encoding subunits of complexes II and IV, contain NRF-2-recognition sites [38]. Breen and Jordan [14] demonstrated complex regulation of expression of CcOVb subunit by several transcription factors. According to these investigators, upstream stimulatory factor (USF) stimulates expression CcOVb via binding to an initiator element in the promoter of the gene encoding this protein, and p300/CBP acts as a coactivator in this process. Another regulatory factor, Yin Yang 1 (YY1), modestly stimulates CcOVb expression but greatly reduces the level of USF-dependent activation of CcOVb transcription. Finally, the transcription factor Sp1 represses both the USF2- and YY1-dependent effects on CcOVb expression, indicating competition among Sp1, YY1, and USF2 (Table 6.1).
Recent evidence suggests that for optimal function of complex I and the ETC at large, the action of transcription factor signal transducer and activator of transcription 3 (STAT3) is required: STAT3 knockout specifically reduced activities of complexes I and II [43]. Activation of STAT3-responsive genes includes interleukin-6-mediated phosphorylation of STAT3. In mouse primary pro-B lymphocytes, knockout of tyrosine kinase Tyk2 that phosphorylates STAT3 has been shown to result in insufficient basal mitochondrial O2 consumption; impaired function of complexes I, III, and IV; and decreased levels of cellular ATP [44]. Thus, Tyk2-catalyzed phosphorylation of STAT3 is required for proper expression and functioning of ETC.
Another aspect of nucleus-related modulation of mitochondria function is tissue-specific expression of subunits of complex IV—cytochrome c oxidase (COX). Several COX subunits are expressed in the heart and skeletal muscle as specific H isoforms: VIa-H, VIIa-H, and VIII-H in beef but only VIa-H and VIIa-H in human. Human heart and skeletal muscle express H isoforms of COXVIa and COXVIIa, and VIa and VIIa subunit promoters contain muscle-specific MyoD-recognition sequence for regulatory myogenic regulatory transcription factor MyoD [15, 45]. Importantly, the heart-specific subunit COXVIa-H but not the lung-specific subunit COXVIa-L is regulated by ADP: ADP binding to the heart enzyme stimulates COX activity [46]. This regulatory cardiac-specific mechanism may be essential for survival of the individual under conditions of increased heart rate. In this situation, ATP consumption in the heart leads to increased ADP concentrations in the mitochondrial matrix, with subsequent increase of the respiratory rate and ATP synthesis, independently of respiratory control. In addition, various hormones (e.g., thyroid hormone, adrenal steroid hormones) have been reported to induce specific COX isoform expression.
Fourteen subunits of ATP synthase are encoded by nDNA. The γ subunit of the catalytic F1-ATPase subcomplex was found to be expressed as two tissue-specific isoforms derived from a single gene by alternative splicing of exon 9, with H isoform prevalent in cardiac and skeletal muscle [47]. Three isoforms of c subunit of the membrane Fo subcomplex (P1, P2, and P3) are encoded by 3 different genes and also show different expression patterns in regard to tissue specificity, physiological regulation, and hormone regulation [48].
Genes encoding α and β subunits of the catalytic F1-ATPase subcomplex contain binding sites for regulatory nuclear factors Sp1, OXBOX, and NRF-2.
Interestingly, transcription and translation of mtRNA is controlled by several nDNA-encoded transcription factors, such as mitochondrial transcription factors TFAM, B1 (TFB1M), and B2 (TFB2M); mitochondrial transcription termination factor (mTERF); mitochondrial translation-initiation factor 2 (mtIF-2); and mitochondrial elongation factors (mtEF) Tu, Ts, and G. Expression of TFAM, TFB1M, and TFB2M is coordinated with that of nDNA-encoded OXPHOS proteins by nuclear transcription factors NRF-1 and NRF-2 [38].
Pyruvate dehydrogenase complex (PDHC) catalyzes the physiologically irreversible oxidative decarboxylation of pyruvate to acetyl-CoA, thereby linking the glycolysis to the TCA cycle. Short-term regulation of this key enzyme complex is achieved by reversible phosphorylation/dephosphorylation (see below). The regulatory components of PDHC, kinases (PDHKs), and phosphatases (PDPs) are also subjects of nuclear differential transcriptional controls, needed for long-term tuning of PDHC activity to maintain cellular glucose homeostasis in different nutritional and disease states.
In starvation, diabetes, and hyperthyroidism, PDHK4 and PDHK2 have been shown to be upregulated in several tissues, whereas PDP1 and PDP2 are downregulated [49–51]. As a result, PDHC activity decreases to conserve glucose for gluconeogenesis. Recently, it has been suggested that glucocorticoids regulate transcription of PDHK4. According to this model, the transcription factor Sp1 and CCAAT box binding factor (CBF) are responsible for the basal level of PDHK4 expression. Glucocorticoids, which are elevated in starvation and diabetes, bind and activate glucocorticoid receptor (GR), which interacts with the glucocorticoid response element (GRE) in the promoter of PDHK4 and recruits p300/CBP. Histone acetylation by p300/CBP enhances the recruitment of the transcriptional machinery and thus enhances transcription of PDHK4. FOXO factors bind to insulin response elements (IRE) of PDHK4 promoter and cooperate with GR. Retinoic acid-bound nuclear retinoid X receptor (RXR)/retinoic acid receptor (RAR) heterodimer further stabilizes transcriptional initiation complex to fully activate the PDHK4 gene expression—Akt phosphorylates/inactivates FOXO and causes its relocation from nucleus to cytosol [20].
In liver, kidney, and heart, the expression of PDHK4 is also activated by FAs through peroxisome proliferator-activated receptor (PPAR) α [ 32, 33] and can be stimulated by PPARγ coactivator [52]. Thus, when FAs are used as an energy source, this regulatory mechanism directs available pyruvate through gluconeogenesis, to provide glucose for the brain and other neural tissues.
The regulated expression of genes encoding mitochondrial FA utilization enzymes plays an important role for energy production via fatty acid β-oxidation (FAO) pathway in accordance with diverse dietary and physiologic conditions. Ligand-activated transcription factors that are known to control the expression of many genes involved in cellular FA import and oxidation are PPARs. Thus, PPARα null mice show reduced rates of cardiac FA utilization due to diminished expression of genes encoding FAO enzymes. On the other hand, cardiac-restricted upregulation of PPARα activates mitochondrial FAO in parallel with a reduction in glucose uptake and oxidation [53]. In agreement with this, recent observations have shown that the PPARα regulatory pathway is deactivated in cardiac hypertrophy and hypoxia, two pathological conditions characterized by reduced FAO and increased dependence on glucose as a fuel source [54]. Moreover, in the diabetic heart, which relies primarily on FAO for energy production, the activity of the PPARα gene regulatory pathway is increased.
In the heart, activation of PPARα increases the expression of genes involved in three major steps in the cellular FA utilization pathway: FA transport and esterification, FA mitochondrial import, and mitochondrial β-oxidation. FAO-mediated enzymes known to be regulated by PPARα include very-long-chain (VLCAD), long-chain (LCAD), and medium-chain (MCAD) acyl-CoA dehydrogenases and 3-hydroxyacyl-CoA dehydrogenase (Table 6.1).
Ligand activation of PPAR leads to its heterodimerization with RXR and subsequent binding of PPARα/RXR heterodimer to the specific DNA response element in the promoter region of the target genes. Extensively characterized PPARα-regulated promoter is the promoter of the MCAD gene that is known to contain a nuclear hormone receptor response element (NRRE-1), which can be activated by several nuclear hormone receptors, including PPARα/RXR heterodimer [34, 36].
The first natural ligand identified for PPARα was the arachidonic acid derivative leukotriene B4 [55]. The major natural ligands for PPARα in the heart are long-chain (C14–C22) FAs. In ligand-binding assays, saturated and mono- and polyunsaturated FAs have been shown to bind and activate PPARα [56–58]. Additional level of PPARα-dependent regulation of target genes involves activation of transcription by recruitment and interaction with transcriptional coactivators. Upon binding to ligand, PPARα changes conformation allowing its C-terminal coactivator interaction domain to become available for interactions with coactivators, such as SRC-1 [59, 60], CBP/p300 [60], PBP/TRAP220 [61], and PGC-1 [40].
Analysis of promoter regions has shown that two nuclear-encoded key TCA-cycle dehydrogenases, α-ketoglutarate dehydrogenase (KGDH) and succinate dehydrogenase (SDH), have potential for complex regulation by nuclear transcription factors. KGDH is an oligomeric complex comprising the three catalytic components. Regarding the first component, α-ketoglutarate dehydrogenase (E1), only the regulation of the gene, encoding the heart isoform of the E1, has been characterized—the α-ketoglutarate induces the promoter activity of this gene. Interestingly, glutamate facilitates the effect of α-ketoglutarate, while alone is inactive [62]. The human dihydrolipoyl succinyltransferase (E2) gene contains glucocorticoid-responsive and CREB-binding elements, as well as a binding site for transcription factor Sp1 [21]. The third human KGDH-related gene, encoding dihydrolipoyl dehydrogenase (E3), has also been shown to contain the cAMP-response element-like site, and its basal transcription depends on CREB [23]. Expression of the KGDH components E1 and E3 is often increased as a compensatory response under conditions causing KGDH inactivation such as ethanol-dependent stress or potassium deficiency [63, 64]. In animal model of increased physiological activity (exercise-trained rats), a twofold increase in cardiac E2 subunit of KGDH has been demonstrated [65].
SDH is the only enzyme that participates in both the TCA cycle and the ETC (as complex II, see above). Four subunits of SDH are encoded by nuclear genes, SDHA–SDHD. In the promoter region of all four SDH genes, NRF-1 binding sites are found. In addition, there are NRF-2 responsive elements in genes SDHB–SDHD; Sp1-binding sites in genes SDHA, SDHB, and SDHD; and TFAM-binding sites in SDHA and SDHB (Table 6.1) [10].
A growing body of evidence indicates that high-energy molecules such as ATP and phosphocreatine may be compartmentalized in cells. Gajewski et al. [66] have studied cell lines with impaired OXPHOS function, caused by mutated mitochondrial ATP synthase (T-to-G transversion at position 8,993 in the ATPase 6 gene) or by defective respiratory chain complexes containing mtDNA-encoded subunits (mutation associated with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes, MELAS). Using the ATP reporter luciferase localized in the nucleus, they demonstrated collapse of ATP levels in the mitochondria and nucleus of mutant cells. These findings suggest that in patients with oxidative energy dysfunction, loss of ATP in the nucleus, because of critically low output of ATP from mitochondria, may have potentially severe consequences in cells with high metabolic demand and high oxidative rates (e.g., myocytes, neurons), resulting in the impairment of crucial cellular functions, such as nuclear import and DNA transcription. These findings are in agreement with observations that import of histone proteins into neonatal cardiomyocyte nuclei depends mainly on mitochondrial ATP synthesis and trafficking of high-energy phosphoryls by phosphotransfer enzymes, such as adenylate kinase and creatine kinase [67].
Mitochondria/Cytosol Interactions
Fatty Acids
Substrate availability to the heart affects mitochondrial metabolism. For example, elevated glucose in cardiomyocytes stimulates glycosylation of mitochondrial ETC proteins leading to impaired mitochondrial OXPHOS [68]. Under conditions with elevated free FA (FFAs) (characteristics of diabetes mellitus Type 2), glucose oxidation decreases due to inhibition of pyruvate dehydrogenase [69]. This switch to FAs as the primary myocardial energy source occurs via regulation of expression of PDHK4 [70]. This kinase is responsible for the inhibitory phosphorylation of PDHC, located in the mitochondrial matrix, and plays an essential role in aerobic energy metabolism, specifically in transport of pyruvate into the mitochondria and pyruvate oxidation.
Within the cell, FAs first become esterificated to fatty acyl CoA by fatty acyl CoA synthetase. For mitochondrial FAO initiation, the cytoplasmic long-chain fatty acyl CoA must first be transported into the mitochondrial matrix. There is a carnitine-dependent transport system responsible for transport of fatty acyl CoA into mitochondrion. This transport system involves three enzymes: carnitine palmitoyltransferase I (CPT-I), carnitine acyltranslocase, and carnitine palmitoyltransferase II (CPT-II). CPT-I converts fatty acyl CoA to fatty acyl carnitine, which crosses the MIM and enters the mitochondrial matrix via diffusion facilitated by carnitine acyltranslocase. Within the mitochondria, CPT-II transfers fatty acyl from carnitine back to CoA. Carnitine then diffuses back across the membrane into the inter-mitochondrial membrane space by a carnitine acyltranslocase, whereas fatty acyl CoA enters β-oxidation process (Fig. 6.1).
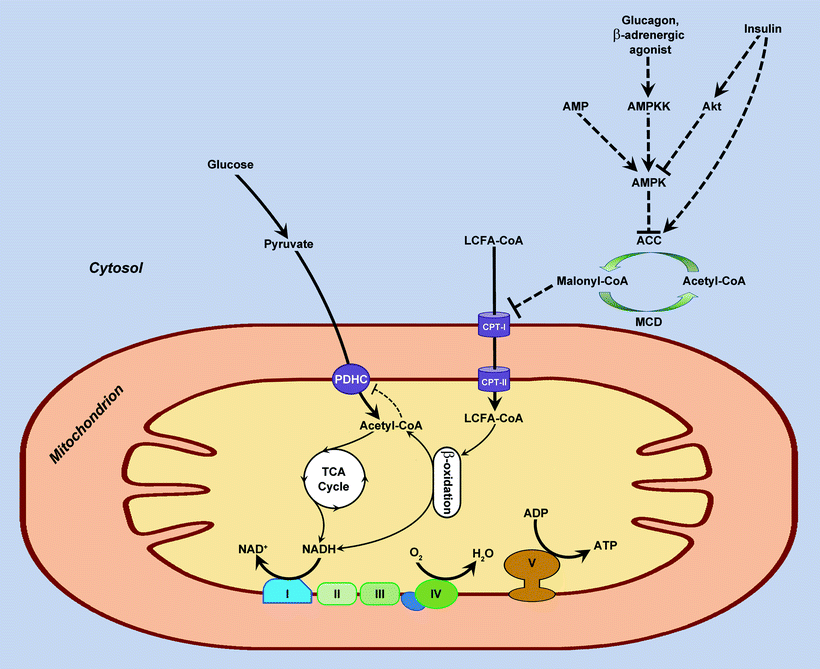
Fig. 6.1
Fatty acid metabolism and malonyl-CoA-related regulation of fatty acid oxidation. Fatty acid enters cell either via diffusion or via CD36/FATP transporter and becomes esterified in the cytosol to long-chain fatty acyl coenzyme A (LCFA-CoA). Special shuttle system transfers fatty acyl from cytosol to mitochondrion. First, acyl group of LCFA-CoA is transferred to carnitine via carnitine palmitoyltransferase-I (CPT-I). The acylcarnitine is then shuttled into the mitochondrion and converted back to LCFA-CoA by carnitine palmitoyltransferase-II (CPT-II). LCFA-CoA enters the fatty acid β-oxidation cycle, producing reducing equivalent NADH and acetyl-CoA. The latter enters the tricarboxylic acid (TCA) cycle to produce more reducing equivalents, NADH and FADH2 (only NADH is shown). Reducing equivalents are oxidized by the complexes I, II, III, and IV of the respiratory chain with an energy transformed into a form of a transmembrane proton gradient that is used, in turn, by the ATP synthase (complex V) to produce ATP from ADP and phosphate. Malonyl-CoA is an endogenous inhibitor of CPT-I, and therefore it is a negative regulator of fatty acid β-oxidation. Malonyl-CoA is generated in cytosol from acetyl-CoA by acetyl-CoA carboxylase (ACC), whereas it is degraded back to acetyl-CoA via decarboxylation by malonyl-CoA decarboxylase (MCD). ACC is regulated allosterically by AMP and by phosphorylation catalyzed by AMP-activated protein kinase (AMPK). Fatty acid metabolism has reciprocal relationship with the metabolism of another major energy source, glucose. Acetyl-CoA, NADH, and FADH2 produced by fatty acid β-oxidation decrease glucose/pyruvate oxidation via the inhibition of pyruvate dehydrogenase complex (PDHC). From the other side, when high level of glucose/pyruvate takes place, an increased cytosolic acetyl-CoA serves as a source of malonyl-CoA, an inhibitor of fatty acid β-oxidation (see above). Metabolic conversions are shown by solid lines; regulatory effects are shown by dashed lines. AMPKK, AMPK kinase
Of the three enzymes participating in mitochondrial FA uptake, CPT-I is rate limiting and is subject of potent regulation. A key molecule responsible for allosteric inhibition of CPT-I is malonyl-CoA. Thus, changes in malonyl-CoA concentration can alter mitochondrial uptake of FA and consequently the rate of FAO in the mitochondria. The concentration of malonyl-CoA is controlled by its turnover, in which acetyl-CoA carboxylase (ACC) catalyzes its synthesis and malonyl-CoA decarboxylase (MCD) controls its degradation. The activity of ACC is in turn determined by the activity of AMP-activated protein kinase (AMPK). This serine/threonine kinase is a real “fuel sensor,” because it responds to metabolic stresses by increasing AMP/ATP and creatine/phosphocreatine ratios. AMPK is a heterotrimer, consisting of a catalytic α subunit and regulatory β and γ subunits. Activation of AMPK involves phosphorylation of Thr-172 on the catalytic α subunit [71]. Several kinases are known to phosphorylate and activate AMPK in mammalian cells, including protein kinase LKB1, the calmodulin-dependent protein kinase kinase, and TGF-β activated kinase 1 (TAK1) [71, 72]. The myosin light chain kinase is a potential kinase that activates AMPK during cardiac ischemia [73]. Once activated, AMPK phosphorylates and inhibits ACC, which in turn relieves CPT-I from inhibition by malonyl-CoA, and FAO increases (Fig. 6.1) [74, 75]. In addition, AMPK can directly activate MCD [76], and high activity of MCD is associated with increased FAO [77]. In contrast, MCD inhibitors increase level of malonyl-CoA that results in decreased FAO and increased glucose oxidation up to tenfold in isolated beating heart [78].
Besides serving as mitochondrial energy substrate, long-chain FFAs exert a series of effects on the mitochondrial bioenergetics affecting the MIM and allosteric regulation of mitochondrial enzymes. Changes in cellular FFA levels can be physiological (fasting, exercise) or pathological (obesity, diabetes, ischemic/postischemic heart).
Proton-pumping efficiency of mitochondrial OXPHOS complex III (cytochrome bc 1 complex) is known to be decreased at alkaline pH, probably because of depressed protonation of amino acid residue(s) at the input mouth of the pump [79]. FFA (arachidonic acid [AA], in particular) at sub-micromolar concentrations facilitates protonation of these critical residues, thus acting as an internal protonophore. It turns out that AA rescues ΔpH-dependent depression of the proton-pumping activity of complex III and is essential for ETC functioning [80, 81].
At micromolar concentrations, FFA may give rise to a so-called mild uncoupling effect. This effect is due to a cyclic movement of undissociated FFAs with the release of protons into the alkaline mitochondrial matrix and subsequent efflux of the FFA anions mediated by mitochondrial anion carriers, such as ANT, aspartate/glutamate, and dicarboxylate carriers [82–84]. The strongest uncoupling activity was found for C12–C16 saturated FAs and for longer cis-unsaturated FAs [85]. Mild uncoupling plays a preventive role by decreasing ROS generation, when mitochondrial transmembrane potential is high [86]. Importantly, FFA accumulation results in both a stronger uncoupling and inhibition of ETC enzymes. Under these conditions, generation of ROS increases and induces oxidative stress response, which can lead eventually to cell necrosis.
Adenine Nucleotides
Mitochondrial ANT controls the flux of ADP and ATP. It is a critical factor in modulating the OXPHOS rate through respiratory control, depending on the metabolic conditions of the tissue. Specifically, ANT regulates the OXPHOS activity of the newborn heart as well as the adult heart under conditions of high workload and hypertrophy [87–89]. ANT is responsible for an adequate supply of ADP to mitochondria, required for the ATP synthase to generate ATP and for the transfer of ATP synthesized in the mitochondria to the sites of utilization (e.g., myofibril, membrane pumps). Furthermore, ATP and ADP are significant allosteric effectors of TCA-cycle enzymes.
Two ANT isoforms are expressed in the heart—ANT1, encoded by gene containing TATA, CCAAT, and OXBOX sites in the promoter region, whereas ANT3, a “housekeeper” isoform, encoded by gene with no regulatory sites in the promoter being identified.
In addition to regulating adenine nucleotide pools, ANT also interacts with voltage-dependent anion channel (VDAC), hexokinase, and mitochondrial creatine kinase [90, 91]. In vitro studies have shown the functional coupling of peripheral kinases to ANT. Atractyloside, a specific inhibitor of ANT, when added to ATP-loaded vesicles with hexokinase–VDAC–ANT complex or VDAC–creatine kinase–ANT complex, inhibits glucose-6-phosphate formation from external glucose (hexokinase activity) or creatine phosphate generation from external creatine (creatine kinase activity) [92]. Tight coupling of peripheral kinases to ANT provides an additional level of regulation for the cell energy metabolism. First, hexokinase-catalyzed glucose phosphorylation, at the expenses of intramitochondrial ATP, activates mitochondrial OXPHOS. Second, ADP produced by creatine kinase is immediately removed by ANT from the extramitochondrial compartments, which balances the reaction between creatine and ATP toward creatine phosphate production and higher free energy of cytosolic ATP (Fig. 6.2).
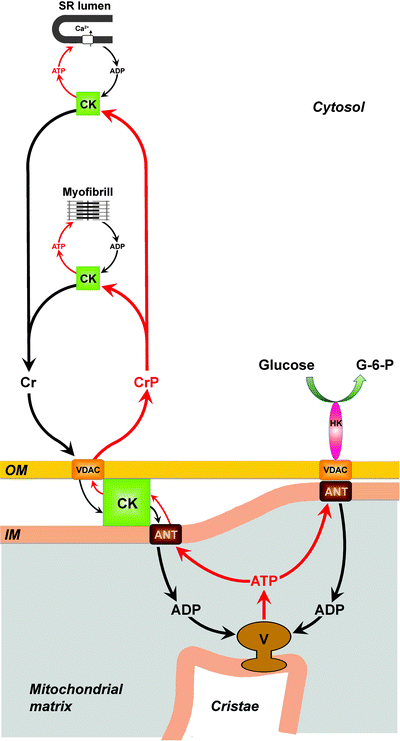
Fig. 6.2
Metabolite channeling mitochondrial and cytosolic microdomains. Mitochondrial creatine kinase (CK) is localized in the intermembrane space of mitochondria. In the inner mitochondrial membrane (IM), CK functionally interacts with adenine nucleotide translocator (ANT). In the outer mitochondrial membrane (OM), CK interacts, in a Ca2+-dependent manner, directly with mitochondrial voltage-dependent anion channel (VDAC). ATP generated by mitochondrial ATP synthase (complex V) is transported through the IM by ANT. In the intermembrane space, CK catalyzes ATP/creatine phosphate (CrP) high-energy phosphoryl transfer. Then, CrP is channeling from mitochondria toward the cytosol via VDAC and feeds into the cytosolic CrP pool. In the cytosol, CrP is available to cytosolic CKs for generation of ATP at the various locations. The cytosolic CK isoenzymes can associate with cellular structures, in close proximity to ATP-requiring processes, and locally use cytosolic CrP to regenerate ATP. Shown is cytosolic CK associated with two major cardiac ATP-consuming processes; CK binds to sarcomeric M-band of myofibrils and regenerates the ATP hydrolyzed by the actomyosin ATPase at contraction; CK is functionally coupled to Ca2+-ATPase pump in the sarcoplasmic reticulum (SR) where it provides the pump with ATP for sequestration of Ca2+. The biological work through activity of cytosolic CKs triggers cytosolic ADP–ATP turnover leading to the increase of cytosolic creatine (Cr). Cytosolic Cr is transphosphorylated in mitochondria by matrix-generated ATP into CrP by mitochondrial CK, and the resulting ADP is channeled into the mitochondrial matrix by ANT. As a result, Cr causes an increase of intramitochondrial ADP and thus stimulates mitochondrial respiration. The contact sites formed by the VDAC–ANT complexes promote oligomerization of hexokinase (HK) leading to activation of the enzyme. The HK bound to the VDAC–ANT complex catalyzes glucose phosphorylation at the expenses of intramitochondrial ATP and thus activates oxidative phosphorylation. ATP and CrP fluxes are shown by red arrows; ADP and Cr fluxes are shown by black arrows. G-6-P, glucose-6-phosphate
ATP utilization in cardiac cells (mainly at the filaments and sarcoplasmic reticulum) is highly balanced with the rate of mitochondrial ATP production. The feedback of the products of ATP hydrolysis, ADP and Pi or creatine, is essential in the regulation of this balance [93, 94]. However, in vivo, the concentrations of ATP, ADP, Pi, and creatine phosphate are maintained in the myocardium in the face of physiological challenges. Currently, two major models have been proposed to explain the cytoplasmic regulatory mechanism(s) of mitochondrial OXPHOS. A metabolite compartmentation model relies on small pools of ADP at the interface of the mitochondria and cytosolic elements, [95] distribution of creatine kinase isoforms in the different compartments of the cell, [96, 97] and “channeling” of ADP at the sarcoplasmic reticulum/mitochondria interface [98, 99]. The second model relies on the main role of Ca2+ in the orchestration of OXPHOS (see below).
Hexokinases
Hexokinases (HKs) play an important role in mitochondria–cytosol cross talk. These enzymes are highly regulated proteins, which can be activated in the myocardium by insulin, hypoxia, or ischemia. They phosphorylate intracellular glucose to maintain the concentration gradient driving myocardial uptake of glucose from the bloodstream and prevent leaking of glucose out of the cell. Under conditions where oxygen availability is limiting to maintain FAO (e.g., increased workload, disease, or injury), glucose phosphorylation by HK is the rate-limiting step in glucose uptake/glycolysis and is subject of regulation to maintain an appropriate level of mitochondrial OXPHOS and cardiac contractility.
Recently, it has been demonstrated that two cardiac HK isoforms, HKI and HKII, translocate to the mitochondria from cytosol within 30 min of either insulin stimulation or ischemia [100]. Binding to mitochondria is mediated by the interaction of an N-terminal domain of HKs with the VDAC on the mitochondrial outer membrane (MOM) and serves a number of functions [101]. First, binding to mitochondria markedly decreases inhibition of HK by its product (glucose-6-phosphate, G-6-P); as a result, HK can continue phosphorylating glucose at high intracellular G-6-P concentrations [102]. Second, mitochondrion-associated HK preferentially utilizes intramitochondrial ATP; thus, glucose uptake is coupled with OXPHOS-generated ATP (Fig. 6.2) [103].
Role of Nitric Oxide
At low [O2] conditions, mitochondria develop a protective mechanism against tissue hypoxia. At low [O2], mitochondrial COX stays in reduced form and is not able to consume nitric oxide (NO•). Elevated NO• resulting from its decreased inactivation by COX activates the soluble guanylate cyclase to produce vasodilatation and therefore to increase the local supply of oxygen.
In addition, NO•-dependent inhibition of mitochondrial respiration at low [O2] results in redistribution of oxygen toward nonrespiratory oxygen-dependent targets, thus regulating oxygen-sensitive pathways.
Hormonal Regulation
In multicellular organisms complex signaling pathways adjust OXPHOS to the individual requirements of various cell types via posttranslational modification of key ETC players. [104] According to traditional views, there are two major regulators of ETC activity and therefore mitochondrial energy and ROS production: (1) the mitochondrial membrane potential (ΔΨm) and (2) allosteric regulation of cytochrome c oxidase by ATP/ADP ratio. Recently, it has been proposed that posttranslational modifications, specifically reversible phosphorylation, may play an essential role in the regulation of OXPHOS and ΔΨm. Phosphorylation sites have been mapped in all mammalian ETC complexes, cytochrome c, and ATP synthase. Among identified phosphorylation sites are tyrosine residues in flavoprotein of complex II [105], catalytic subunits I and II and subunit IV of COX [106–108], cytochrome c [109, 110], and δ subunit of ATP synthase [111].
Many protein kinases and phosphatases are located inside the mitochondria or translocate into the mitochondria upon their activation (Fig. 6.3). Although little is known about the regulatory mechanisms underlying phosphorylation of mitochondrial proteins, several signaling pathways have been shown to induce multiple phosphorylation events in mitochondria, including PDGF [111], EGF [112], and TNF-α [107]. Non-receptor tyrosine kinase Src localizes to mitochondria and can phosphorylate COXII [113].
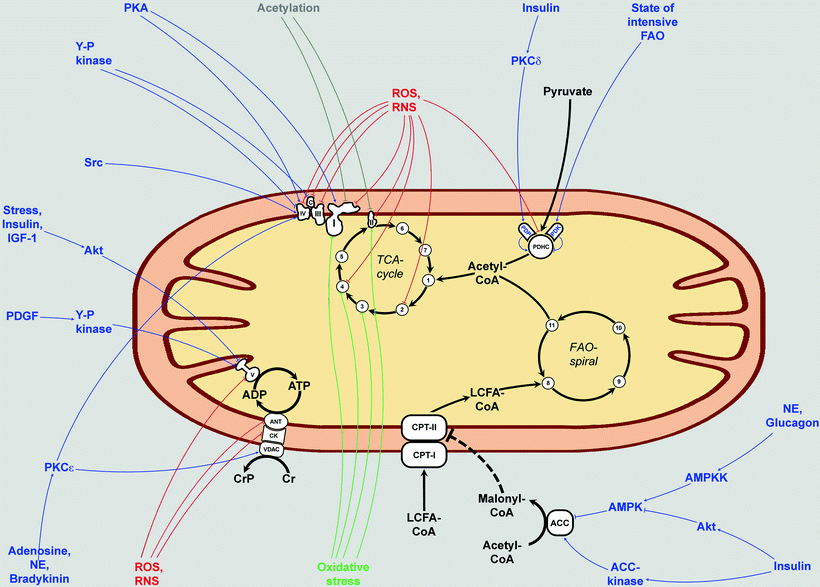
Fig. 6.3
Involvement of extracellular signaling pathways in the regulation of mitochondrial bioenergetics. The major pathways of energy production within the mitochondria are shown: tricarboxylic acid (TCA) cycle, fatty acid β-oxidation (FAO) spiral, electron transport chain (ETC) complexes I–IV and cytochrome c (c), ATP synthase (complex V), and pyruvate dehydrogenase complex (PDHC). Also major mitochondrial ATP–ADP and long-chain fatty acid (LCFA)-transporting systems are depicted (ANT/CK/VDAC and CPT-I/CPT-II, respectively). Metabolic conversions are shown by solid black arrows; regulatory inhibition of CPT-II by malonyl-CoA is shown by dashed black T-ending line. Many of the mitochondrial complexes are subjected to reversible extramitochondrial signal-initiated phosphorylations and dephosphorylations that affect mitochondrial energy-producing function (shown in blue). ROS and RNS that are evoked during oxidative or nitrosative stress can result in oxidative damage to the mitochondria via ROS- and RNS-dependent covalent modifications of proteins (shown in red). Major targets for S-glutathionylations which are also activated during oxidative or nitrosative stress are indicated by green lines. Several components of ETC also can be acetylated by protein acetyltransferases (shown in grey). This process is reversible: major protein deacetylase in mitochondria is sirtuin 3 (not shown). 1, citrate synthase; 2, aconitase; 3, isocitrate dehydrogenase; 4, α-ketoglutarate dehydrogenase; 5, succinyl-CoA synthase; 6, fumarase; 7, malate dehydrogenase; 8, acyl-CoA dehydrogenase; 9, enoyl-CoA hydratase; 10, 3-hydroxyacyl-CoA dehydrogenase; 11, 3-ketoacyl-CoA thiolase. Other abbreviations: ACC acetyl-CoA carboxylase; AMPK AMP-activated protein kinase; AMPKK AMPK kinase; ANT adenine nucleotide translocator; CK creatine kinase; CoA coenzyme A; CPT carnitine palmitoyltransferase; Cr creatine; CrP creatine phosphate; IGF-1 insulin-like growth factor 1; NE norepinephrine; PDGF platelet-derived growth factor; PDK pyruvate dehydrogenase kinase; PDP pyruvate dehydrogenase phosphatase; PKA protein kinase A; PKC protein kinase C; RNS reactive nitrogen species; ROS reactive oxygen species; VDAC voltage-dependent anion channel; Y-P kinase unknown tyrosine kinase. Arrows, stimulatory effects; T-bars, inhibitory effects
It has been reported that upon activation protein kinase C (PKC) isoforms are translocated into the mitochondria. For example, in myeloid leukemia cells, PKCδ activated by phorbol 12-myristate 13-acetate (PMA) translocates to mitochondria with subsequent release of cytochrome c and apoptosis [114]. Similarly, translocation of PKCδ to mitochondria takes place in perfused rat hearts during reperfusion after ischemia [115] and in human breast cancer cells after hydrogen peroxide-induced oxidative stress [116]. In the latter case, PKCδ translocation was accompanied by release of cytochrome c and the loss of ΔΨm. In neonatal cardiomyocytes, another PKC isoform, PKCε, also translocates to the mitochondria following activation by PMA where it binds to COXIV [117, 118]. Association of PKCε with COXIV is induced by hypoxic preconditioning of neonatal cardiomyocytes resulting in phosphorylation of COXIV and stimulation of cytochrome c oxidase activity. Thus, PKCε can enhance myocardial resistance to ischemic injury via modulation of myocardial bioenergetics to increase ATP synthesis [117].
Stimulation of human neuroblastoma and embryonic kidney cells with insulin-like growth factor-1 (IGF-1), insulin, or stress (heat shock) causes phosphorylation and translocation to mitochondria of protein kinase B (Akt) where it phosphorylates the β subunit of ATP synthase and other proteins [119]. Unfortunately, in most cases the specific mitochondrial targets of the translocated protein kinases remain yet to be determined (Fig. 6.3).
Another kinase that has been of particular interest in the regulation of mitochondrial bioenergy-producing function is cAMP-dependent protein kinase (PKA). PKA can be localized to the mitochondrial matrix and/or to the MOM, via interactions with anchoring proteins [120–122]. A number of ETC components have been identified as PKA substrates in vitro (Fig. 6.3) [123–125]. cAMP-dependent phosphorylation of several subunits of complex IV has been observed in an isolated rabbit heart model of ischemia, but the current literature about PKA-dependent effects on mitochondrial OXPHOS and ROS production remains controversial [120, 126].
Recently, Lee et al. [127] reported data on the role of dephosphorylation in cardiac COX activity. In fibroblasts overexpressing constitutively active phosphotyrosine phosphatase SHP2, cardiac COX activity was significantly increased. Similarly, COX activity was elevated in Noonan syndrome patients with activating mutations in the PTPN11 gene encoding SHP2. SHP2 is known to localize to the mitochondrial intercristae/intermembrane space where it can target phospho-sites on cardiac COX subunits or indirectly stimulate Src-dependent COXII phosphorylation.
It appears that the terminal enzyme of the respiratory chain, COX, represents the controlling site of respiration and ATP synthesis in living cells. Therefore, it is not surprising that COX is regulated by reversible phosphorylation. Phosphorylation of subunit I of COX is inhibitory [107], whereas phosphorylations of subunits II and IV increase activity of COX [117, 128]. In general, a very important property—allosteric ATP inhibition—is possible only in phosphorylated COX. Kadenbach et al. [104, 129] hypothesized that under relaxed conditions, phosphorylated COX via allosteric feedback inhibition by ATP keeps mitochondrial ΔΨm at low values to prevent the formation of ROS and maintain efficient OXPHOS. Various stress factors lead to dephosphorylation of COX (probably via activation of a Ca2+-dependent phosphoprotein phosphatase). Without allosteric ATP inhibition of COX under stress conditions, mitochondrial ΔΨm rises to high values with subsequent increase of ROS formation. Chronic stress and elevated ROS levels could lead to cell apoptosis and the generation of degenerative diseases.
The allosteric ATP inhibition of COX highly depends on the concentration of reduced cytochrome c (ferrocytochrome c, CytC 2+)—it occurs at low concentrations (1–5 μM) but is abolished at concentrations above 10 μM of CytC 2+ [130]. As complexes I, II, and III supply COX with CytC 2+, regulation of them can also contribute to the functioning of mitochondrial respiration and COX. In growing C6 glioma cells, OXPHOS is adjusted to energy demand through the cell growth rate, and this adjustment is mainly due to the regulation of the phosphorylation level of complex I subunits. In particular, two small integral membrane proteins belonging to the supernumerary or accessory group of subunits of complex I, NADH dehydrogenase (ubiquinone) 1 β subcomplex subunit 11 (ESSS) and NADH dehydrogenase (ubiquinone) 1 α subcomplex subunit 1 (MWFE), are regulated through phosphorylation. Phosphorylation of ESSS is under the control of the cAMP signaling pathway [131]. Strong regulation by PKA-dependent phosphorylation has been demonstrated for another complex I subunit, NADH dehydrogenase (ubiquinone) iron-sulfur protein 4 (NDUFS4) [123]. In a model system (Chinese hamster mutant cell line missing MWFE), expression of MWFE with serine-55 substituted with glutamate (mimicking the phosphorylated serine) results in failure to assemble the mature complex and hence complete loss of complex I activity [132].
Several of the subunits of the ATP synthase were found to be phosphorylated in the matrix (Fig. 6.3). Extramitochondrial Ca2+ was found to participate in dephosphorylation of the γ subunit of the enzyme. Possibly this dephosphorylation of the γ subunit is partially responsible for the modulation of ATP synthase activity by Ca2+ [133]. Other phosphorylation sites in the ATP synthase complex could also contribute to activity modulation [134, 135]. The identification and functional significance of these protein phosphorylation sites in the ATP synthase are being actively studied in several laboratories.
Besides ETC/ATP synthase, other components of the mitochondrial energy-producing machinery can be targets for hormonal regulation (e.g., PDHC) [70]. The target of regulatory phosphorylation/dephosphorylation is the α subunit of PDH containing three phosphorylation sites: Ser-264, Ser-271, and Ser-203 [136, 137]. These sites can be phosphorylated independently, and each phosphorylation event results in PDH inactivation [138]. In mammals, four isoenzymes of PDH kinase (PDHK) are present with differences in their specific activities, their regulation, and their tissue-specific distribution. PDHK1 and PDHK 4 are highly expressed mainly in the heart, whereas PDHK2 is abundant in many other tissues. All PDHKs can phosphorylate Ser-264 and Ser-271; however, Ser-203 is phosphorylated by PDHK1 only [139, 140]. High levels of pyruvate and a low energy state (high levels of ADP, NAD+, CoA, and Pi) inhibit PDHK leading to the increase in PDHC activity [141]. In addition, PKCδ has been shown to affect the function of PDHK as well as the activity of PDHC [142]. Importantly, two PDH phosphatases (PDP), PDP1 and PDP2, have been detected in the heart; they can dephosphorylate all three phosphorylation sites on the α subunit of PDH. Randle [143] has proposed a classical enzyme regulation model showing that PDH in the mitochondrial matrix is activated by Ca2+-dependent dephosphorylation, mediated by PDP1. Insulin has been shown to induce PKCδ [144], phosphoinositide 3-kinase, and mitogen-activated protein kinase (MAPK) pathways [145] that phosphorylate and activate PDP (Fig. 6.3).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


