Benign Tumors of the Lung
Philip G. Robinson
Thomas W. Shields
Benign tumors of the lung are infrequently encountered. Martini and Beattie171 reported that <1% of the lung tumors resected at Memorial Sloan-Kettering Hospital were benign. Chan and colleagues40 pointed out that the benign lung tumors probably accounted for 2% to 5% of all primary lung tumors. In the older literature, during the 1960s and less so in the 1970s, most of the adenomas were carcinoid tumors or cylindromas, which is another name for adenoid cystic carcinoma. The change in terminology makes it difficult to evaluate the older literature. In this chapter, there is no discussion of either of these lung tumors, since they are not benign. Finally, we do not discuss lymphoid lesions of the lung. Benign tumors may be derived from all cell types present in the lung and may be either parenchymal or endobronchial in location.
No standardized classification of benign lung tumors exists. The authors took the liberty of creating classifications (Table 124-1 for solitary tumors and Table 124-2 for multiple tumors) mostly derived from the World Health Organization (WHO) classification of tumors and including the lung group authored by Travis and colleagues271 and the soft tissue group authored by Fletcher and associates.80 Since the pulmonary hamartoma is the most common of the benign tumors, we elected to discuss this entity first and to discuss the remaining entities in the order in which they appear in the two classification tables of solitary and multiple benign pulmonary tumors (Tables 124-1 and 124-2).
Some of the tumors in this section were considered benign, but in some cases they may recur or metastasize. These tumors include pleomorphic adenoma, myoepithelioma and adenomyoepithelioma, solitary fibrous tumor, inflammatory myofibro- blastic tumor, and benign metastasizing leiomyoma.
Historical Perspective
Steele253 published a review of the resection of 887 pulmonary nodules. The nodules could be subdivided as follows: granulomas (53.4%), malignant tumors (35.6%), hamartomas (7.3%), miscellaneous tumors (2.5%), and pleural or chest wall tumors (1%). Shortly thereafter, Aletras and colleagues5 discussed 16 “benign” lung tumors that consisted of the following: 8 adenomas (subdivided into 90% carcinoid tumor and 10% pleomorphic adenoma), 5 hamartomas, 1 schwannoma, 1 ateriovenous fistula, and 1 fibroma (probably a solitary fibrous tumor). Arrigoni and coworkers11 reported 130 cases of benign lung tumors from the Mayo Clinic. The tumors they reported could be classified as follows: hamartoma (76.9%), benign fibrous mesothelioma (probably solitary fibrous tumor) (12.3%), xanthoma and inflammatory pseudotumors (5.4%), and other (5.4%). In these early series, the pulmonary hamartoma is clearly the most common benign tumor.
Since the early papers mentioned above, other series of benign lung tumors have appeared. Between 1974 and 1988, Mitsudomi and associates181 resected 36 benign tumors or tumor-like lesions out of 721 thoracotomies. The tumors were hamartoma (58%), inflammatory pseudotumor (25%), and sclerosing hemangioma (16%). Otani and colleagues206 reported, in their 20-year experience with benign lung tumors, that the most common ones were hamartoma (54%), sclerosing hemangioma (18%), bronchogenic cyst (13.6%), leiomyoma (9%), and adenoma (4.5%). In conclusion, the hamartoma is the most common benign tumor of the lung. Allen,6 in his review of rare solitary benign lung tumors, agreed that the hamartoma was the most common. More recently, Smith and colleagues250 reviewed their experience of benign lung disease in 140 out of 1,560 patients from January 1995 to December 2002 who underwent resection for focal pulmonary lesions that were thought to be malignant. The lesions could be classified as follows: granulomatous inflammation in 91 patients (65%), hamartoma in 17 patients (12%), pneumonia or pneumonitis in 14 patients (10%), fibrosis in 5 patients (4%), and other in 13 patients (9%). The category of other lesions includes abscess, aspergilloma, amyloid, bronchogenic cyst, carcinoid tumorlet, clear cell tumor, inflammatory pseudotumor, meningioma, mesothelial hyperplasia, necrosis, and rheumatoid nodule. Again, the pulmonary hamartoma remains the most common benign solitary tumor.
Hamartoma
A pulmonary hamartoma is a benign neoplasm composed of varying proportions of cartilage, fat, connective tissue, and smooth muscle with respiratory epithelium. This lesion is known by a variety of names, including chondroid hamartoma, benign mesenchymoma, hamartochondroma, chondromatous hamartoma, adenochondroma and fibroadenoma of the lung, and possibly fibrolipochondroma. According to the historical perspective above, this is the most common benign lung tumor. Bateson20 described this lesion as a benign true neoplasm of fibrous connective tissue of the bronchi that included passively entrapped respiratory epithelium. Whyte and Donington,282
having completed a thorough review of hamartomas from a clinical perspective, point out that the endobronchial lesions contain chromosomal abnormalities that suggest a clonal origin for these tumors.
having completed a thorough review of hamartomas from a clinical perspective, point out that the endobronchial lesions contain chromosomal abnormalities that suggest a clonal origin for these tumors.
Table 124-1 Benign Solitary Tumors of the Lung | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 124-2 Benign Multiple Tumors of the Lung | |||||||
|---|---|---|---|---|---|---|---|
|
Incidence
Khouri and associates137 reported that hamartomas represent 4% of all solitary pulmonary nodules. Rarely, multiple lesions are observed, as one of us (T.W.S.) encountered and as recorded by Bennett and associates.23 Minama and coworkers180 described a hamartoma that was growing diffusely along the bronchial tree and required a pneumonectomy. The rare cystic pulmonary hamartoma has been reported in the literature at least nine times. A few of these reports were by Jackson,123 Demos,55 and Miura183 and their colleagues. The remaining hamartomas are endobronchial in location. Le Roux158 recorded an incidence of 8% of endobronchial hamartomas in 27 patients. Gjevre and associates,96 however, noted an incidence of only 1.4% in 215 cases of hamartoma seen at the Mayo Clinic. The true incidence undoubtedly lies somewhere between these two percentages.
Clinical Features
Hamartomas are most common in the middle-aged adult, although no age group is exempt. Arrigoni and associates11 reported that pulmonary hamartomas are observed twice as often in men as in women. Only 3 of their 102 hamartomas were endobronchial. Ge and coworkers92 described 67 patients from 1970 to 1997 with pulmonary hamartomas. There were 38 men and 29 women, a male-to-female ratio of 1.3:1. The patients’ ages ranged from 21 to 82 years, with a mean age of 47 years. The peak incidence was between 40 and 60 years. Thirty-nine percent of the patients were symptomatic, with hemoptysis, cough, phlegm, or chest pain. Most patients with peripherally located lesions are asymptomatic. Only those few patients who have an endobronchial lesion have symptoms that include cough, hemoptysis, and, frequently, repeated or persistent pulmonary infection.
In contrast, Gjevre and associates96 found 215 patients with pulmonary hamartomas at the Mayo Clinic between 1976 and 1992. They noted that only 3% were symptomatic and that the ratio of men to women was 2:1. Slow growth of the lesion may
be observed (see Fig. 124-2 C,D), but the doubling time usually is well above that of malignant lesions (see Chapter 96). Hansen and colleagues110 reported that the size of the hamartoma could increase by an average of 3.2 ± 2.6 mm per year.
be observed (see Fig. 124-2 C,D), but the doubling time usually is well above that of malignant lesions (see Chapter 96). Hansen and colleagues110 reported that the size of the hamartoma could increase by an average of 3.2 ± 2.6 mm per year.
Most recently, over 1971 until 2002, Lien and colleagues162 described 62 pulmonary hamartomas in 61 patients. The ages ranged from 20 to 77 years with a mean of 56.9 years. The ratio of men to women was 2.8:1. Forty-one (67.2%) patients were asymptomatic, with 20 patients (32.8%) being symptomatic. The symptomatic ones complained of persistent cough, chest tightness, and repeated respiratory tract infections. The four with endobronchial tumors were symptomatic. The majority of parenchymal lesions were asymptomatic (72%). The lesions were equally distributed between all the lobes of the lungs.
Radiology
Ninety percent of pulmonary hamartomas present as solitary peripheral masses (Fig. 124-1 A,B). Radiographically, the peripheral lesion, most often located in the lower lung fields, appears as a smooth and well-circumscribed mass; at times, the margins appear lobulated or, more specifically, bosselated. The usual size is 1 to 2 cm, but larger lesions occasionally are observed. Calcifications have been noted in 10% to 30% of these lesions. On computed tomography (CT) examination, however, Ledor and associates154 found identifiable calcification in <5% of these tumors. When present, the calcification occurs most often in a diffuse or popcorn distribution. This calcification can be seen on high-resolution CT (HRCT) scans (Fig. 124-2). Siegelman and colleagues245 also reported that fatty tissue was identified in 50% of the hamartomas evaluated by CT. In the areas of the fat content, the CT number [Hounsfield unit (HU)] is often low (40–120) and is of diagnostic significance when at least eight voxels are involved. Alternating areas of fat and calcification (HU >175) are also diagnostic. It is now accepted that the presence of a fat density identified by the HRCT scan (Fig. 124-3) in a peripheral solitary lesion is strong presumptive evidence that the lesion is a benign hamartoma and excision can be deferred.
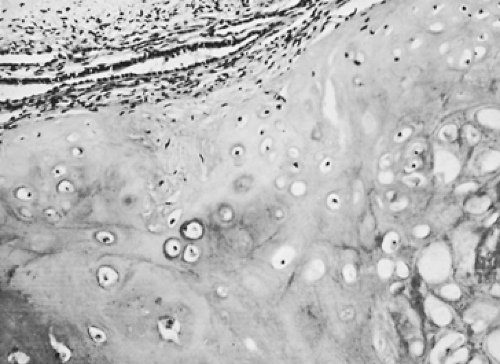 Figure 124-1. Photomicrograph of a hamartoma. Note the predominance of cartilage cells. |
The endobronchial lesions are undetectable radiographically except that distal parenchymal lung changes (e.g., atelectasis, obstructive pneumonia, or abscess formation) may suggest an obstructing, endobronchial lesion.
Pathology
On gross examination, hamartomas have a gray lobulated or bosselated surface. On cut section, they have a gray appearance (Fig. 124-4). There may be some gritty yellow areas that represent calcifications. On microscopic examination, hamartomas are composed of lobulated masses of mature cartilage with other mesenchymal elements that include adipose tissue, smooth muscle, bone, and fibrovascular as well as fibromyxoid tissue (Fig. 124-5). Frequently there are cleft-like spaces between the lobules of cartilage that are lined by respiratory epithelium. Pelosi and colleagues209 reported that these tumors contain estrogen, progesterone, and androgen receptors. The androgen receptors were present in 53% of the hamartomas but were confined to men and present in the fibromyxoid component. The estrogen and progesterone receptors were in 90% of the hamartomas and were present in both men and women. The progesterone receptors were in the fibromyxoid component and the estrogen receptors were in the epithelial cells.
In the cases of Lien and colleagues,162 the tumors ranged in size from 0.2 to 5 cm in greatest dimension with a mean size of 1.8 cm. On gross examination, the tumors were well-circumscribed, firm nodules. On microscopic examination, they were composed of lobules of cartilage surrounding fibromyxoid stroma and adipose tissue. The majority of the hamartomas were described as chondromatous (44 cases or 91.6%). The remaining ones consisted of lipochondromatous, fibrous, and fibroleiomyomatous types.
Treatment
Prior to treatment, a diagnosis should be established. If the diagnosis of hamartoma is suspected from the standard radiographic or CT studies but doubt still remains as to the diagnosis, a needle aspiration biopsy is indicated, particularly in a patient who is a poor candidate for a major operation. Hamper and colleagues107 reported that percutaneous, transthoracic needle aspiration biopsies yield diagnostic information in 85% of hamartomas. Care must be taken in aspirating these peripheral lesions because of their firm consistency. The aforementioned authors reported a 50% incidence of postaspiration pneumo- thorax, which is twice the incidence after biopsy of other peripheral nodules. One may suspect that the lesion is a hamartoma when fibromyxomatous tissue, which stains metachromatically with Giemsa or Wright’s stain, and fragments of low columnar epithelium are present. When fragments of cartilage are present cytologically, the aspiration is diagnostic of a hamartoma. Azua Blanco and colleagues15 performed a fine-needle aspiration (FNA) on a pulmonary hamartoma. They found spindle and stellate cells with a fibromyxoid background, which enabled them to establish a diagnosis before surgery. The histologic examination of the aspiration has a higher diagnostic yield than cytologic examination of the aspirated specimen. Cartilage is more often demonstrated by standard histologic examination of the aspirated material.
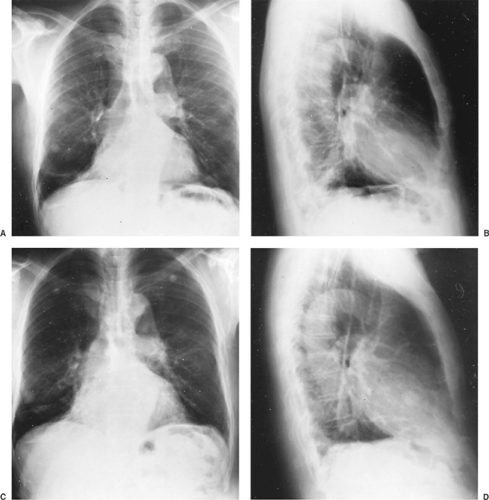 Figure 124-2. Hamartoma demonstrated on posteroanterior (A) and lateral (B) radiographs of the chest, presenting as a peripheral mass in the middle lobe. Posteroanterior (C) and lateral (D) radiographs of the chest of the peripheral hamartoma show slow growth of the mass over a 4-year period. |
Bronchoscopy and biopsy are indicated in any patient with pulmonary symptoms: cough, hemoptysis, repeated pulmonary infection, or atelectasis. Endoscopy is not an essential diagnostic step in patients with a peripheral lesion.
If a prethoracotomy diagnosis has not been made, removal by video-assisted thoracoscopic surgery (VATS) of a peripherally located suspected hamartoma is an acceptable approach. Thoracotomy and excision may be indicated at times. Hamartomas may be small and difficult to visualize. Powell217 and Eichfeld65 and their colleagues have described inserting a microcoil or a spiral wire into the lesion percutaneously under CT guidance. They found that it was easier to locate the lesion and it probably decreases the rate at which VATS is converted to thoracotomy. The least possible amount of normal pulmonary tissue should
be excised. At times, when a suspected hamartoma is palpated within the lung, the mass is readily moved and may be advanced to just beneath the visceral pleural surface. Incision of the pleura and enucleation of the mass can then be readily carried out. Any fixation of the mass within the parenchyma of the lung necessitates a standard resection (wedge resection), segmentectomy, or even at times a lobectomy. A pneumonectomy should be avoided if at all possible. Recurrence after excision of a hamartoma is practically unknown. A second, separate primary hamartoma rarely occurs later.
be excised. At times, when a suspected hamartoma is palpated within the lung, the mass is readily moved and may be advanced to just beneath the visceral pleural surface. Incision of the pleura and enucleation of the mass can then be readily carried out. Any fixation of the mass within the parenchyma of the lung necessitates a standard resection (wedge resection), segmentectomy, or even at times a lobectomy. A pneumonectomy should be avoided if at all possible. Recurrence after excision of a hamartoma is practically unknown. A second, separate primary hamartoma rarely occurs later.
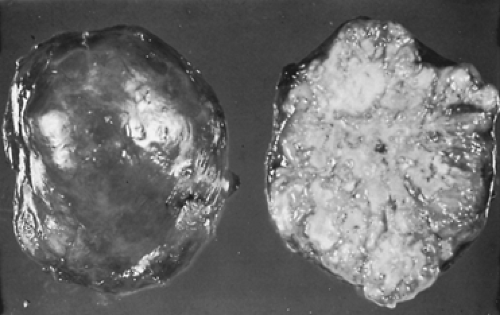 Figure 124-3. Gross specimen of a resected hamartoma, showing typical bosselated appearance. |
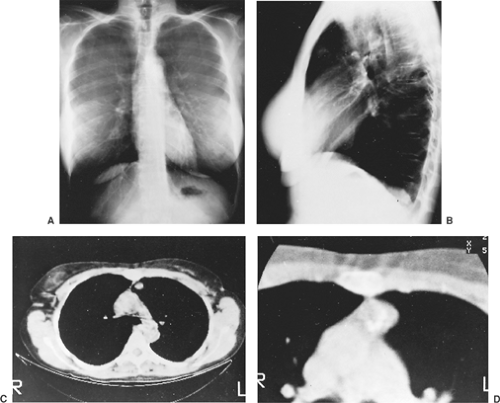 Figure 124-4. A: Posteroanterior radiograph of calcified lesion overlying aortic knob on the left. B: Lateral radiograph shows the lesion to be in the anterior segment of the left lung. C: CT scan showing calcification within the mass. D: Enhanced CT scan revealing popcorn-like calcifications in the mass, typical of a hamartoma. |
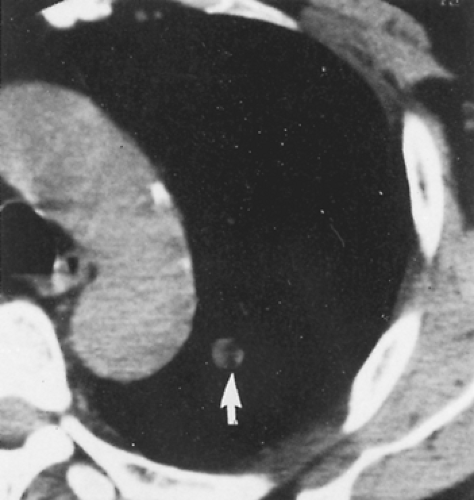 Figure 124-5. Thin-section CT scan demonstrating a 1-cm pulmonary nodule with fat density material within (arrow). Lesion has appearance consistent with a hamartoma. *From Swensen SJ et al. An integrated approach to evaluation of the solitary pulmonary nodule. Mayo Clin Proc 1990;65:173. With permission.) |
The treatment of endobronchial hamartomas has evolved over the years; at present, most of these lesions are best managed by endoscopy and laser ablation, as recorded by Cosio and coworkers.48 This approach was successful in 38 patients, most of whom (86.1%) had distal pulmonary obstruction, although 4 patients had subsequent local recurrence of the tumors. In 3 of these, a second laser ablation was curative. In another 5 patients of the total of 43 patients in the aforementioned series, standard thoracotomy and pulmonary resection were required for the removal of the tumor and the destroyed distal pulmonary parenchyma, as one of us (T.W.S.) and Lynn242 described in 1958. Tomos and colleagues269 described a bronchoplasty technique for the removal of endobronchial hamartomas.
When the diagnosis was known in a patient with a peripherally located hamartoma, Nili and associates199 reported that the patient could be observed without surgical intervention. de Rooij and coworkers59 concur with this suggestion, but they believe that if the mass is >2.5 cm, it should be removed. We believe that clinical judgment should be the final determinant as to whether lesions >2.5 cm should be removed. Minimal growth over time may be noted. Unless the growth rate becomes excessive, excision remains unnecessary. In a patient in whom the diagnosis has been established by histologic evaluation of a needle biopsy, the necessity of resection, except under unusual circumstances, can be questioned.
Malignancy Associated with Hamartoma
There have been several reports of malignancy occurring in a hamartoma. Hayward and Carabasi111 presented a patient in whom an adenocarcinoma was believed to have developed from a hamartoma. Basile and associates19 recorded a sarcoma that developed promptly at the site of a resected benign hamartoma. Neither of these events is convincing on close scrutiny, and no real evidence exists that either tumor actually arose from an underlying hamartoma. One may be as critical of these reports as were Hayward and Carabasi111 of the 12 cases of possible malignancy they reviewed in their own publication. However, of interest in this regard is the possible potential of malignant change. Okabayashi and colleagues203 reported a giant hamartoma associated with a high production of carbohydrate antigen 19–9. The significance of this is unknown, but the source of the antigen was demonstrated to be the epithelial component and not the mesenchymal component. Further investigations, such as those of Fletcher and coworkers,81 might provide insight into any possible malignant potential of these two components of this normally benign biphasic tumor.
Karasik and associates129 reported that a bronchial carcinoma (synchronous or metachronous) was identified 6.3 times more often in patients with a hamartoma than would be expected in the normal population. They suggested that an etiologic relationship was present. Van den Bosch and colleagues,276 however, who identified 6 synchronous and 5 metachronous bronchial carcinomas in a series of 154 patients with hamartomas (an incidence of 7%), believed the association was essentially coincidental. In a more recent series of 65 patients with hamartomas, Ribet and associates220 recorded that 3 patients had an associated bronchial carcinoma, a 6.6-fold increase in the number of cases normally expected. These authors came to the same conclusion that there was an etiologic relationship present, as had Karasik and colleagues.129 However, in the other series of a total of 598 patients with hamartomas, the rate of occurrence of a lung cancer was only 5.8%. The question of the nature of the relationship between these two lesions remains unresolved.
Other Solitary Benign Tumors
Since the hamartoma is the most common of all the solitary benign tumors, it is discussed first. Thereafter, the tumors are discussed in the order in which they are listed in Tables 124-1 and 124-2. Table 124-3 shows those tumors that are mainly endobronchial. Benign tumors of epithelial and soft tissue origin as well as the miscellaneous tumor are rare. Many of these tumors may be either endobronchial or peripheral in location but generally have a greater predilection for one of the two locations. The symptomatology depends on whether a bronchus is irritated or a bronchial lumen is occluded partially or completely by an endobronchial lesion. The peripherally located tumors usually are asymptomatic.
Table 124-3 Solitary Tumors of the Lung That May Be in an Endobronchial Location | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Epithelial Tumors
Papilloma
The subject of pulmonary papillomas is confusing, because multiple squamous papillomas may occur in the upper respiratory tract and larynx and may eventually involve the tracheobronchial tree and lungs. The pulmonary papillomas, both squamous and glandular, discussed in this section are solitary and involve the tracheobronchial tree and lungs. Occasionally, there is some overlap between the squamous papillomas of recurrent respiratory papillomatosis and solitary squamous papillomas. Older discussions and classifications described a disease that begins in the larynx and pharynx with latter involvement of the tracheobronchial tree and lungs.
According to Derkay and Wiatrak,58 upper respiratory papillomatosis is due to infection with the human papillomaviruses (HPV) types 6 and 11. The squamous papillomas may recur and they may involve the tracheobronchial tree and the lungs. When the squamous papillomas recur, the disease becomes known as recurrent respiratory papillomatosis (RRP). Recurrent respiratory papillomatosis can be divided into two forms: a juvenile onset form and an adult onset form. The juvenile form usually has its onset in the teen years, but it can start as early as the first year of life. This lesion is discussed in Chapter 81. The adult form tends to present in the third and fourth decades. Pulmonary squamous papillomas should be viewed a separate entity from recurrent respiratory papillomatosis.
Squamous Papilloma
A papilloma is a benign epithelial neoplasm composed of multiple fronds of papillary squamous epithelium; it usually has a thin fibrovascular core. The majority of papillomas are exophytic, but a few inverted ones have been described. An inverted papilloma grows downward toward the underlying stroma and is thus called inverted rather than exophytic.
Flieder and associates82 comprehensively reviewed the subject of solitary pulmonary papillomas—including the squamous, glandular, and mixed papillomas—with 14 cases of their own and 27 cases from the literature. They described 27 patients with squamous cell papillomas, with 5 being their own patients. The patients ranged in age from 28 to 74 years with a median age of 54 years. There were 23 men and 4 women; 6 had a history of smoking. Five of their patients were symptomatic, with hemoptysis, recurrent pneumonia, and wheezing; three of their five patients had radiographic abnormalities, including one with a hilar mass and two with postobstructive pneumonia and bronchiectasis. In the 27 cases, the papillomas were equally distributed between the left and the right lungs. The tumors ranged in size from 0.7 to 9.0 cm, with a median size of 1.5 cm in greatest dimension. On microscopic examination, the lesions were composed of papillary arborizing connective tissue stalks lined by keratinizing or nonkeratinizing squamous epithelium. Of the 5 patients, 1 had a tumor that was positive for HPV types 6 and 11 but negative for 16, 18, 31, 33, and 51. Two other patients were tested and were negative for all of the previously listed types of human papillomavirus. All 5 patients were alive and well with the follow-up periods ranging from 3 months to 16 years.
HPV is probably the cause of most of these lesions. Popper and colleagues216 demonstrated that HPV types 11 and 6 were associated with benign papillomas, whereas types 16 or 18, sometimes in combination with type 31, 33, or 35, were found in papillomas of patients who developed squamous cell carcinoma. Katial and associates131 reported an endobronchial squamous papilloma that was positive for types 6 and 11 but negative for 16, 18, 31, 33, and 35. Kawaguchi and coworkers134 reported a solitary squamous papilloma of the right upper lobe that contained HPV type 11. Lam and associates152 reported HPV 6b in an atypical squamous papilloma of the trachea.
Iwata and coworkers121 reported an inverted Schneiderian papilloma in the right lower lobe of the lung. This tumor is noteworthy because few inverted papillomas are reported in the lung, and it was associated with elevated serum levels of carcinoembryonic antigen and squamous cell carcinoma–associated antigen. Once the lesion was removed, the levels returned to almost normal. HPV types 6, 11, 16, and 18 were not detected in the tumor.
Miura and colleagues182 pointed out that treatment should be conservative, but patients may require further surgery if they develop a malignancy. They suggested that if the lesion is limited to a small area, it could be treated with photodynamic therapy, yttrium-aluminum-garnet laser, or both; these patients should be followed. These aforementioned lesions are usually removed endoscopically, or occasionally, a bronchotomy or sleeve resection may be necessary. When irreversible parenchymal damage distal to the lesion is present, surgical resection of the destroyed lung tissue also is required.
Glandular Papilloma
A glandular papilloma is a benign papillary tumor lined by ciliated or nonciliated columnar epithelial cells. A synonymous term is columnar cell papilloma. According to Flieder and colleagues,82 these are extremely rare endobronchial tumors that occur at a median age of 68 years with no predilection for sex. Patients may present with obstructive symptoms, such as wheezing or hemoptysis. On microscopic examination, the tumor can have thick arborizing stromal stalks covered by glandular epithelium. These tumors are benign, but they may recur following incomplete resection.
Mixed Squamous Cell and Glandular Papilloma
A mixed squamous cell and glandular papilloma is a benign endobronchial papillary tumor showing a mixture of squamous and glandular epithelium. A synonymous term is transitional cell papilloma, and this is what Colby and colleagues47 called it in their description. Flieder and colleagues82 point out that these are extremely rare tumors with only seven cases reported in the world literature. The tumors have an equal sex distribution and the median age is 64 years. Patients may have obstructive symptoms. The histology shows fibrovascular cores that lined by both squamous and glandular epithelium. Complete resection appears to be curative.
Adenoma
Alveolar Adenoma
An alveolar adenoma is a benign, well circumscribed peripheral lung tumor. It may be mistaken for a lymphangioma. Yousem
and Hochholzer295 reported six patients with alveolar adenomas. They were mostly women and ranged in age from 45 to 74 years. Most of the lesions were found on routine chest radiography. Radiographically, Fujimoto and colleagues86 describe alveolar adenomas as peripheral, well-circumscribed solitary nodules. Magnetic resonance imaging (MRI) shows the nodules to have a cystic space with fluid and thin rim enhancement. Cavazza38 and Hartman108 and their colleagues also described alveolar adenomas as showing a cystic component. Halldorsson and associates105 report a positron emission tomography (PET) scan and the lesion showed no uptake. On excision, the tumors averaged 2 cm in diameter and were easily shelled out from the adjacent pulmonary parenchyma. On histologic examination, the tumors are composed of spaces lined by a low cuboidal epithelium with underlying connective tissue stroma that may be myxoid. Cavazza and colleagues38 describe adipose tissue in the lesion as well as the mesenchymal cells immunostaining for S-100. Halldorsson and associates105 report that the epithelial cells immunostain for cytokeratin CK7 and CK20 as well as thyroid transcription factor 1 (TTF-1). Five of the six patients were alive and well at the end of a 12-month follow-up period, with one patient lost to follow-up. Saito and colleagues230 described a case of an alveolar adenoma.
and Hochholzer295 reported six patients with alveolar adenomas. They were mostly women and ranged in age from 45 to 74 years. Most of the lesions were found on routine chest radiography. Radiographically, Fujimoto and colleagues86 describe alveolar adenomas as peripheral, well-circumscribed solitary nodules. Magnetic resonance imaging (MRI) shows the nodules to have a cystic space with fluid and thin rim enhancement. Cavazza38 and Hartman108 and their colleagues also described alveolar adenomas as showing a cystic component. Halldorsson and associates105 report a positron emission tomography (PET) scan and the lesion showed no uptake. On excision, the tumors averaged 2 cm in diameter and were easily shelled out from the adjacent pulmonary parenchyma. On histologic examination, the tumors are composed of spaces lined by a low cuboidal epithelium with underlying connective tissue stroma that may be myxoid. Cavazza and colleagues38 describe adipose tissue in the lesion as well as the mesenchymal cells immunostaining for S-100. Halldorsson and associates105 report that the epithelial cells immunostain for cytokeratin CK7 and CK20 as well as thyroid transcription factor 1 (TTF-1). Five of the six patients were alive and well at the end of a 12-month follow-up period, with one patient lost to follow-up. Saito and colleagues230 described a case of an alveolar adenoma.
Bohm and colleagues26 as well as Oliveira and associates204 have each described an additional case of alveolar adenoma. Both groups believe that it is a distinct lung neoplasm. Burke and coworkers27 reviewed 17 cases and performed a variety of immunohistochemical stains on the tumors. They concluded that alveolar adenomas were benign neoplasms consisting of an admixture of alveolar epithelium, mostly type 2 pneumocytes, and septal mesenchymal tissue, fibroblasts, or fibroblast-like cells. Oliveira and colleagues204 speculate that the lesion is derived from a primitive mesenchymal cell with the capacity to differentiate toward a type 2 pneumocyte lineage. In contrast, Bohm and associates26 believe the neoplasm is derived from a benign proliferation of both the type 2 pneumocytes and the septal mesenchyme. Cavazza and coworkers38 performed microsatellite analysis on the epithelial and mesenchymal components of an alveolar adenoma and found alterations of the epithelial cells but not the mesenchymal ones. These findings suggest that the tumor is derived from the epithelial cells.
Papillary Adenoma (Type II Pneumocyte Adenoma or Clara Cell Adenoma)
A papillary adenoma is a rare benign papillary neoplasm that is thought to arise from a multipotential stem cell that differentiates toward type II pneumocytes, Clara cells, or ciliated respiratory epithelial cells. Synonyms for this lesion include Clara cell adenoma, bronchiolar papilloma, bronchiolar adenoma, type II pneumocyte adenoma, and papillary adenoma of type II pneumocytes. Spencer and colleagues251 described two cases. Fantone75 and Noguchi201 and their associates described papillary adenomas of the lung that had ultrastructural differentiation toward type 2 pneumocytes (lamellar bodies) and Clara cells (membrane-bound, electron-dense granules). Hegg,112 Sanchez-Jimenez,234 Fukuda,87 Mori,189 and Dessy60 and their colleagues have all added additional cases to the literature.
The patients range in age from 2 months to 60 years, and the tumor may occur in either gender. The lesions are usually detected in asymptomatic patients as a result of mass radiologic screening in the periphery of the lung. On gross examination, the tumors are usually described as well-demarcated white nodules in the pulmonary parenchyma. Microscopic examination shows a papillary architecture with prominent fibrovascular cores. The epithelial cells, lining the cores, are predominantly cuboidal with basal nuclei and eosinophilic cytoplasm. Sheppard and colleagues240 point out that the surface cells immunostain with thyroid transcription factor 1 (TTF-1). Ultrastructurally, Clara cells and type II pneumocytes are identified. The differential diagnosis includes alveolar adenoma, papillary bronchioloalveolar carcinoma, papillary variant of sclerosing hemangioma, papillary variant of carcinoid tumor, and metastatic carcinoma. Resection appears to be curative, with all of the patients surviving for at least 2 to 10 years. Mori and coworkers,189 using morphometry with 12-dimensional cluster analysis, found a resemblance of some of the cells to type II pneumocyte adenocarcinoma, but their patient was alive with no evidence of recurrence at 3 years. Dessy and colleagues60 described two cases of this lesion with infiltrative features. They also found two other cases in the literature with infiltrative features. They proposed changing the name to peripheral papillary tumor of undetermined malignant potential. Infiltrative lesions should be followed closely.
Adenoma of Salivary Gland Type: Pleomorphic Adenoma
A pleomorphic adenoma is a benign tumor showing both epithelial and connective tissue differentiation. These tumors contain glands and myoepithelial cells that are usually set in a cartilaginous stroma and are also known as benign mixed tumors. A carcinoma arising in a pleomorphic adenoma is known as carcinoma ex pleomorphic adenoma. Jin and Park,125 Ang,7 and Tanigaki265 and associates have reported individual cases of pulmonary pleomorphic adenomas.
Sakamoto and colleagues231 reported one case of pleomorphic adenoma in the lung and reviewed six other reported cases. The patients ranged in age from 47 to 74 years, with an average age of 57 years. Both sexes were equally affected by these tumors. The clinical symptoms included pneumonia and cough; one patient was asymptomatic. Moran and colleagues184 described eight patients with pleomorphic adenomas in which two were malignant. They concluded that size at presentation, extent of local infiltration and mitotic activity were the most reliable prognostic features. Moran185 summarized the findings in 16 patients with pleomorphic adenomas. His patients ranged in age from 35 to 74 years; most were women. Hara and associates106 described the radiologic findings. On chest radiograph, the tumor was a well-circumscribed partly lobulated nodule. On unenhanced CT, the tumor was sharply marginated without calcifications. The mass was enhanced heterogeneously. On MRI, the mass showed low to intermediate signal intensity of T1-weighted images and heterogeneous intermediate intensity on T2-weighted images. The tumors could be either endobronchial or parenchymal in location, but no predilection occurred for a particular lung or segment. On microscopic examination, the pulmonary tumors do not have as prominent a cartilaginous stroma as do the salivary gland tumors. Treatment is surgical excision. These patients require long-term follow-up. Some lesions seem to have the ability to recur and metastasize, and they are difficult to identify histologically.
Tracheal pleomorphic adenomas have been reported. Most of these are single case reports, including those of Kim,139 Baghai-Wadji,16 and Aribas10 and their colleagues. The three patients ranged in age from 8 to 42 years, with two males and one female. All three patients were symptomatic and thought to have asthma and all presented with dyspnea. Two of the tumors were in the trachea and one involved the carina and right bronchus. In two of the cases, the tumors ranged from 1.5 to 2 cm in greatest dimension. The histology revealed glands in a chondromyxoid stroma. Surgery is the treatment of choice. As with the pulmonary pleomorphic adenomas, these patients should also receive long-term follow-up, because some lesions seem to have the ability to recur and metastasize.
Adenoma of Salivary Gland Type: Mucous Gland Adenoma
A mucous gland adenoma is an extremely rare benign exophytic tumor of the bronchus that is derived from the mucous glands of the bronchus. The tumor must be composed of cystic glands, be superficial to the cartilaginous plate, be in the bronchus, and have some normal bronchial seromucous glands. The tumor is also known as mucous gland cystadenoma, bronchial cystadenoma, bronchial adenoma arising in mucous glands, mucous cell adenoma, polyadenoma, adenomatous polyp, and adenoma of mucous gland type.
Mucous gland adenoma has been described by Weinberger and associates278 as well as by Gilman,95 Weiss and Ingram,279 Kroe and Pitcoc,149 Emory and associates,68 and Edwards and Matthews.64 England and Hochholzer69 reported 10 additional cases. Their patients ranged in age from 25 to 67 years, with a mean of 52 years. Historically, this lesion tends to occur twice as often in men as in women, but these authors found a slight predominance in women. The symptoms are cough, fever, recurrent pneumonia, and hemoptysis. The chest radiograph may also show obstructive pneumonitis, postobstructive atelectasis, and, on rare occasion, a solitary peripheral lesion. Kwon and colleagues150 reported the CT findings in two patients, which consisted of a well-defined mass with an air meniscus sign or abutting the bronchus, suggesting an intraluminal location. The lesion occurs equally between the right and left sides and more often is found in the major bronchi of the middle and lower lobes. On gross examination, the tumors varied in size from 0.8 to 6.8 cm, with a mean of 1.8 cm. The tumors projected into the lumen of the bronchus. They were usually encapsulated by a thin membrane and easily separated from the bronchus. The cut surface is cystic with mucus within the cystic space.
Endoscopically, they appear as firm pink masses with intact overlying epithelium (Fig. 124-6). Histologically, they are composed of numerous small mucus-filled cysts lined by well-differentiated mucous epithelium (Fig. 124-7). The major differential diagnosis is low-grade mucoepidermoid carcinoma. Even though these lesions rarely have a stalk, they can be completely removed endoscopically by curettage, cryotherapy, or laser ablation, as reported by Ishida and colleagues.119 Thoracotomy and surgical resection are indicated only when distal lung has been destroyed or endoscopic removal is contraindicated or incomplete. Complete removal of these tumors endoscopically or surgically results in a permanent cure. Takeda and colleagues263 performed a left upper sleeve lobectomy on one for a complete cure.
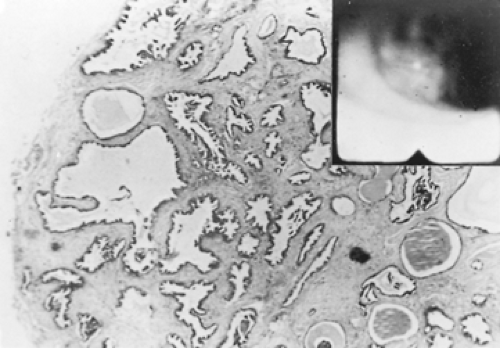 Figure 124-6. Low-power photomicrograph of a mucous gland adenoma. Inset shows the endoscopic appearance. |
Adenoma of Salivary Gland Type: Oncocytoma
An oncocytoma is an extremely rare benign exophytic tumor of the bronchus that is composed of oncocytic cells. The tumor is also known as oxyphilic adenoma or oncocytic adenoma. Fechner and Bentinck77 have described the pulmonary oncocytoma. In addition, Santos-Briz,236 de Jesus,56 Tahiro,261 Genechten,93 and Burrah28 and associates as well as Fernandez and Nyssen,78 Tesluk and Dajee,268 and Laforga and Aranda151 have described pulmonary or tracheal oncocytomas, with all of these papers being single case reports.
The patients in the above reported cases ranged in age from 16 to 75 years with a median age of 40 years and a slight male predominance. Most of the patients were asymptomatic, but those who were symptomatic complained of cough, hemoptysis, chest pain, and dyspnea. The chest radiographs may show obstructive pneumonitis with an infiltrate or a solitary peripheral lesion. The case reported by Laforga and Aranda151 had multiple nodules. The solitary lesions were randomly distributed in the lungs. On gross examination, the tumors were well circumscribed and
varied in size from 1.5 to 3.5 cm in greatest dimension. The cut surface was yellow tan to reddish brown to pink. On microscopic examination, the tumors have ovoid cells with small uniform nuclei and abundant finely granular eosinophilic cytoplasm. On electron microscopy, the cytoplasm shows mitochondrial hyperplasia. The treatment is surgical resection.
varied in size from 1.5 to 3.5 cm in greatest dimension. The cut surface was yellow tan to reddish brown to pink. On microscopic examination, the tumors have ovoid cells with small uniform nuclei and abundant finely granular eosinophilic cytoplasm. On electron microscopy, the cytoplasm shows mitochondrial hyperplasia. The treatment is surgical resection.
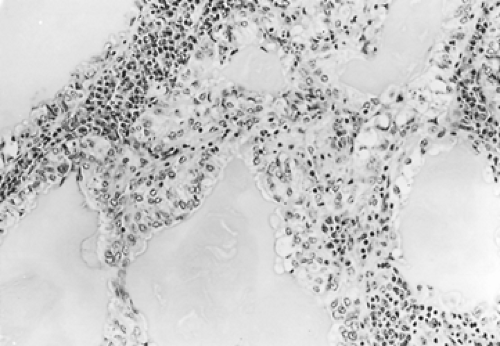 Figure 124-7. High-power photomicrograph of a mucous gland adenoma consisting of cysts of various diameters lined by columnar mucous cells. A chronic inflammatory reaction separates the tubules. |
Adenoma of Salivary Gland Type: Myoepithelioma and Adenomyoepithelioma
The existence of this entity is confusing because of the terminology. Yousem and Nicholson297 consider the terms adenomyoepithelioma, myoepithelioma, epithelial-myoepithelial tumor, epimyoepithelial carcinoma, epithelial–myoepithelial tumor of unproven malignant potential and malignant mixed tumor comprising epithelial and myoepithelial cells to be synonyms with epithelial–myoepithelial carcinoma, which is viewed as a low-grade carcinoma. Chang and colleagues41 recently described a pneumocytic adenomyoepithelioma, which is discussed at the end of this section.
Myoepithelial cells are flat cells that lie between the epithelial cells of a gland and the basement membrane. They are usually found in the salivary glands and are thought to have contractile properties. The older concept of myoepitheliomas was of a benign tumor. Kilpatrick and Limon138 also view these lesions as low-grade malignancies and grouped them with the parachordoma and mixed tumors of soft tissue. The tumors are discussed in this section because they have traditionally been viewed as benign.
Strickler and coworkers254 described a pulmonary myoepithelioma, and Tsuji273 and Pelosi210 and their associates described adenomyoepitheliomas or, as Pelosi and colleagues210 preferred to call them, pulmonary epithelial–myoepithelial tumor of unproven malignant potential (PEMTUMP). They prefer this term because at present these tumors appear benign.
The myoepithelioma described by Strickler and coworkers254 was an incidental finding on a chest radiograph in a man in his 60s. On gross examination, the lesion was 3.3 cm in greatest dimension, had tan-yellow-white surfaces, and had well-demarcated margins. On microscopic examination, it was composed of spindle cells that immunostained for S-100 and actin but not keratin. On electron microscopy, the tumor contained filaments consistent with myofilaments. Cagirici and colleagues29 described a myoepithelioma in a 54-year-old woman. She presented with exertional dyspnea, cough, and intermittent pleuritic chest pain. A chest radiograph showed a peripheral 2-cm mass in the left lower lobe. Grossly, the tumor was white-gray with well-demarcated margins. Microscopically it consisted of uniform spindle shaped cells that immuno- stained for S-100 and smooth muscle actin but not desmin, synaptophysin, or CD34. The patient underwent a wedge resection and is alive and well with no evidence of recurrence at 2 years.
Pelosi and associates210 reported a PEMTUMP and reviewed the six cases in the world’s literature, including the one described by Tsuji and coworkers.273 In the review, the patients ranged from 47 to 66 years of age, with an average age of 56 years. Tumors were present in four women and three men and ranged in size from 1.3 to 16 cm. The cut surfaces were tan-yellow-white and the tumors were randomly distributed throughout both lungs. Radiographically, the lesions were solid nodules. On microscopic examination, the tumors showed a biphasic pattern composed of glands (epithelial cells) and spindle cells (myoepithelial cells). The epithelial cells stained strongly for keratin and weakly for S-100. In contrast, the myoepithelial cells stained strongly for S-100 and actin but weakly for keratin. Overall, the patients did well, but some were lost to follow-up. The longest follow-up was 36 months. None of these patients are known to have died from their disease. Surgery appears curative, but follow-up is advised.
Chang and associates41 described five cases of a new pulmonary tumor that they call pneumocytic adenomyoepithelioma. They view it as a tumor that shows epithelial myoepithelial and pneumocytic differentiation. Their five patients were all women and ranged in age from 52 to 63 years. The history was available for three patients. Two were asymptomatic with the lesion being incidentally discovered on a chest radiograph. The other patient complained of chest pain and dyspnea. The tumors did not show a preferred location and they measured between 0.8 and 2.6 cm in greatest dimension. Microscopically, all five tumors showed a double layered glandular formation and all were close to a small caliber airway. The epithelial cells showed immunostaining for keratin, epithelial membrane antigen (EMA), and thyroid transcription factor-1 (TTF-1). In contrast, the myoepithelial cells showed immunostaining for smooth muscle actin, S-100 and p63, but were negative for keratin. The treatment is surgical. The follow-up period is up to 6.5 years with no recurrence; however, one patient had multiple bilateral nodules and has been followed for only 5 months.
Mucinous Cystadenoma
According to Colby and colleagues,47 a mucinous cystadenoma is a “unilocular cystic lesion whose fibrous wall is lined by well-differentiated, presumably benign columnar mucinous epithelium.” This lesion was first described by Sambrook Gowar233 and later by Dail,52 Kragel,148 Dixon,63 and Roux227 and their colleagues; Graeme-Cook and Mark102 and Dail52 have further described this entity. These lesions occur in both men and women, who are usually in their 50s and 60s and are smokers. Most of these tumors are discovered as asymptomatic masses on routine chest radiography. The mass is usually located at or toward the periphery of the lung.
On gross examination, the mass is a unilocular cyst filled with clear gelatinous material. On microscopic examination, a fibrous cyst wall is lined by mucinous epithelium. Occasionally, the wall is thinned, and the mucin extravasates into the adjacent pulmonary parenchyma. The cysts should be examined completely because they may have areas of borderline malignancy or adenocarcinoma. The differential diagnosis ranges from bronchogenic cysts to bronchoalveolar carcinoma. The treatment for these lesions is complete resection, and the prognosis of the benign tumors is excellent.
Mann and associates167 reported the local recurrence of a mucinous cystic tumor of borderline malignancy 4 years after its initial resection. They suggested that a mucinous cystic tumor with any histologic identification of evidence of even early malignant features should undergo a lobectomy as the procedure of choice rather than a more limited excision. Matsuo and colleagues173 also reported a recurrent mucinous cystadenoma in a 56-year-old woman who had had a partial resection of the lesion 20 years before. After a 2-year follow up, she has no evidence of a recurrence.
Soft Tissue Tumors
Adipocytic Angiomyolipoma
Angiomyolipoma is benign tumor of adipose tissue that contains blood vessels, adipose tissue, and smooth muscle. Marcheix and colleagues168 reported one in the lung of a 63-year-old asymptomatic woman who presented with a chest radiograph that showed a nodule in the right lower lobe. A CT scan showed a nonenhancing nodule with a fat-like density and no calcifications. A PET scan showed a focus of intense tracer location in the right lower lobe. The lesion was resected. On microscopic examination, the lesion was unencapsulated and composed of mature fat with thick-walled blood vessels and smooth muscle cells. Immunohistochemically, these tumors stain with desmin and HMB-45. The patient had no history of tuberous sclerosis or lymphangioleiomyomatosis. These tumors are usually found in the kidneys of patients with tuberous sclerosis.
Lipoma
Lipomas are benign tumors of mature adipose tissue, which may occur in either an endobronchial or a parenchymal location. Lipomas arise most often from the wall of the tracheobronchial tree (80%). These lesions are more common in men than in women. They may cause obstruction with pulmonary complications. Bango18 and Yokozaki291 and their colleagues have urged CT examination to determine the extent of pulmonary involvement. They also suggest bronchoscopic laser vaporization of the tumor as the treatment of choice, although a local resection by bronchotomy or sleeve resection may be required.
Muraoka and associates193 reported 64 cases of endobronchial lipomas from the Japanese literature. Fifty patients were men and 14 were women and they had a mean age of 60 years and a deviation of 11.4 years. The majority of the patients were symptomatic (75%), with the symptoms consisting of sputum, cough, hemoptysis, fever, and dyspnea. Forty of the lipomas were in the right lung and 23 in the left lung. Sixty-one of the lipomas were found in the first three subdivisions of the bronchial tree, and radiographic findings were present in 78% of the patients. The tumors ranged in size from 0.3 to 6.5 cm in greatest dimension. On microscopic examination, the tumors were composed of mature adipose tissue. Surgical procedures—including pneumonectomy (4), lobectomy (24), bilobectomy (8), and bronchotomy (4)—were required in 57.9%. Bronchoscopic removal by Nd:YAG laser (17), electrosurgical resection (5), or a combination of both (5) was carried out in the other 42% of patients. Bronchoscopic removal is preferred whenever possible.
Civi and associates43 reported a case of a peripheral lipoma and reviewed the world literature where they found eight additional cases. There was a male predominance, with seven men and two women; the ages ranged from 44 to 71 years. One patient was asymptomatic and the remaining eight had symptoms that consisted of sputum, cough, fever, hemoptysis, chest pain, dyspnea, wheezing, and right arm paresthesia. The chest radiograph of their patient showed opacity in the left lower lung; four of the other patients had tumors in the right lung and five in the left, with four of the tumors in the left lower lobe. On gross examination, the tumors ranged from 1.3 cm to 7.8 cm in greatest dimension. On microscopic examination, the tumor should consist of mature fat; however, their tumors contained occasional giant cells with multiple pleomorphic nuclei. These could represent floret-like giant cells, as are seen in pleomorphic lipomas. The majority of patients were treated surgically.
Fibroblastic/Myofibroblastic Pulmonary Microcystic Fibromyxoma
A pulmonary microcystic fibromyxoma is thought to be a benign tumor of the lung composed of myxoid connective tissue with microcyst formation. The only well-known myxoid tumor of the lung is the myxoid variant of a pulmonary hamartoma. Shilo and associates243 described three cases of this new tumor. Their patients consisted of two women and one man, with the women being 45 and 65 years old and the man being 45 years old. All were symptomatic and the lesion was found incidentally, with no evidence of an extrapulmonary primary on evaluation. Chest radiographs showed a nodule. On gross examinations, the nodules varied in size from 1 to 2.3 cm in greatest dimension and were described as brownish to white-tan. On microscopic examination, the tumors were composed of bland stellate cells with the formation of microcysts. No mitosis or necrosis was present. Immunohistochemically, the cells were positive for vimentin but negative for keratin, thyroid transcription factor-1, and various other markers. All of the patients are well, with follow-up ranging from 18 to 72 months. The treatment was surgical removal of the nodules.
Inflammatory Fibrous Polyp
A polyp is a mass of tissue protruding from an epithelial surface that is composed mostly of underlying connective tissue and covered by ciliated columnar epithelium with possible areas of squamous metaplasia. The stalk is usually composed of loose connective tissue with capillaries and an infiltrate of plasma cells, lymphocytes, and eosinophils. On rare occasions, there can be multiple polyps. A synonym is inflammatory endobronchial polyp. Patients may present with symptoms of obstruction, such as wheezing or recurrent infections. Dinçer and colleagues62 point out that some of the etiologic factors include foreign-body aspiration, prolonged mechanical ventilation, asthma, chronic sinusitis, chronic smoke inhalation, and mycobacterial infections. Inflammatory polyps tend to be more common in adults, but they can also involve children. Arguelles and Blanco9 described an asthmatic 10-year-old boy who had multiple bronchial polyps. Roberts and associates222 described multiple endobronchial polyps in a girl with a history of cystic fibrosis who at the age of 12 years underwent a resection. McShane and coworkers175 described three infants and a 12-year-old boy with endobronchial polyps. All of the infants had histories of intubation and McShane and coworkers175 postulated that the polyps may have resulted from irritation of the endotracheal tube or the suctioning of the patients. There was no history of intubation in the 12 year old.
In adults, endobronchial polyps may be secondary to a variety of causes. Some of the less common causes include a case secondary to tuberculosis as described by Nishi and colleagues.200 The mycobacteria were identified in the polyp by special stain. Another case is reported by Asano and associates,12 who describe polyps in a woman with disseminated Mycobacterium intracellulare. In this instance the organisms were not identified in
the polyp by special stains. Naber and coworkers195 described endobronchial polyps in lung transplant patients that were secondary to cytomegalovirus. The virus could be identified in the polyp following immunohistochemical staining. Gamblin and associates91 described a tracheal inflammatory polyp in a 43-year-old man.
the polyp by special stains. Naber and coworkers195 described endobronchial polyps in lung transplant patients that were secondary to cytomegalovirus. The virus could be identified in the polyp following immunohistochemical staining. Gamblin and associates91 described a tracheal inflammatory polyp in a 43-year-old man.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


