Benign and Malignant Neurogenic Tumors of the Mediastinum in Children and Adults
Marleta Reynolds
Thomas W. Shields
Neurogenic Tumors in Infants and Children
Neurogenic tumors occurring in the mediastinum, almost exclusively in the paravertebral sulci, in infants and children most commonly arise from tissues of the autonomic ganglia and only infrequently are of nerve sheath origin. Rarely, a tumor of neuroectodermal origin also may occur in this location. Even less common is a lesion arising from the paraganglionic system.
In the majority of reviews of childhood mediastinal tumors, the neurogenic tumors (most of which as noted are of neuronal cell origin) account for up to 40% of the total number of lesions encountered. However, in the Mayo Clinic experience in children ≤19 years of age, as reported by King and associates,66 the incidence was only 28%. These tumors in infants and children may be readily classified, as shown in Table 196-1. In Table 196-2, the number and types of neurogenic tumors seen over a 24-year period at the Children’s Memorial Hospital of Chicago are listed,102 as are the series reported by Saenz and colleagues107 and Ribet and Cardot.103 Of the 162 tumors in these three series, 60% were malignant. The number of cases and the age groups at the time of presentation reported by Reed and colleagues101 from the Armed Forces Institute of Pathology (AFIP) and from the series of Ribet and Cardot103 are seen in Table 196-3.
Tumors of the Autonomic Ganglia
Tumors of the autonomic ganglia arise from the primitive neural crest cells, which can have tangled cell processes resulting in a pink background of neuropil. These tumors are associated with a greater or lesser amount of fibrovascular stroma. The lesions may be frankly benign (e.g., the ganglioneuroma), malignant to varying degrees (e.g., the ganglioneuroblastoma), or frankly and aggressively malignant (e.g., the neuroblastoma). The latter not only invades locally but is associated with widespread distant metastases. It is believed that all three tumors represent a continuum of a process of maturation: the neuroblastoma being the least mature, the ganglioneuroblastoma more mature with an increasing number of mature ganglion cells present, and the ganglioneuroma a fully differentiated benign lesion. In an attempt to simplify pathologic identification of these tumors, Joshi and associates59 established specific terminology for neuroblastic tumors (Table 196-4).
It is generally estimated that more than 50% of the neurogenic lesions in infants and children are malignant. Most of the malignant lesions are seen in the younger patients, and the benign lesions tend to be observed in the older child or adolescent (see Table 196-3). Neuron-specific enolase may be identified in all these tumors. Although Marangos and Schmechel77 considered this to be a specific marker for neural elements and their tumors, Schmechel111 pointed out in an editorial that this marker may be identified in other tumor cell types and in normal cell lines. Gould and associates42 have noted that synaptophysin may also be identified by immunofluorescence microscopy in all these tumors by the tissue’s reaction to the monoclonal antibody SY-38. These investigators also reported that synaptophysin is readily demonstrated in most neuroendocrine neoplasms and does not seem to occur in nonneuroendocrine cells or neoplasms.
Table 196-1 Mediastinal Neurogenic Tumors in Infants and Children | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Table 196-2 Neurogenic Mediastinal Tumors in Children | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
These tumors are capable of producing norepinephrine and dopamine. The levels of the metabolites of these catecholamines may be measured in the blood or urine. Elevated levels of the degradation products vanillylmandelic acid (VMA) and homovanillic acid (HVA) and total metanephrine are found most commonly. Hinterburger and Bartholomew50 and Siegel and associates,119 among others, reported that elevated levels of urinary VMA and HVA are seen in up to 90% of patients with neuroblastomas. Elevated levels of these substances occur to a lesser extent in the other autonomic nerve tumors as well. Clinical symptoms of the excessive catecholamine production may be present but may not be proportionately related to the measured catecholamine levels.
Voute and colleagues135 reported that tumors originating in the dorsal root ganglia do not usually secrete catecholamines, so the absence of elevated VMA or HVA levels does not rule out the diagnosis of a neuroblastoma. Williams and associates139 reported the production of vasoactive intestinal peptide by some of these tumors. This is thought to be the cause of intractable diarrhea seen in some patients with neuroblastoma.
Neuroblastomas
The neuroblastoma is believed to arise from the pluripotential neural crest cell. The most common cytogenetic abnormality found is a deletion of the short arm of chromosome 1. Maris and associates,80 studying 13 patients with familial neuroblastoma, suggested that the neuroblastoma suppressor gene thought to be located at 1p36 is not the only neuroblastoma suppressor gene. Other common cytogenetic abnormalities include the pressure of homogeneously staining regions and double minutes. Homogeneously staining regions and double minutes correspond to N-myc amplification units. Clinical studies by Seeger and colleagues (112) have shown that N-myc amplification is associated with rapid tumor growth and advanced-stage disease. Morris and associates,90 however, have documented that mediastinal neuroblastomas seldom have N-myc amplification.
Pathology.
Grossly, neuroblastomas are large, lobulated, and soft; they may appear to be pseudoencapsulated. The cut surface is gray-red, and multiple hemorrhagic areas are frequently present. Microscopically, the tumor is composed of small cells with scanty cytoplasm. The nuclei are round to polygonal with a salt-and-pepper chromatin pattern. Characteristically, a circular grouping or pseudorosette formation of cells appears around a fine fibrillar network (Fig. 196-1). The background of the tumor may contain varying amounts of neuropil. Occasional large cells with ganglionic differentiation may be seen. Foci of calcification are commonly present. Rarely, as reported by
Stowens and Lin,125 abundant intracytoplasmic neuromelanin (pigmented neuroblastomas) is present.
Stowens and Lin,125 abundant intracytoplasmic neuromelanin (pigmented neuroblastomas) is present.
Table 196-3 Incidence and Age of Presentation of Neurogenic Mediastinal Tumors in Infants and Children | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Table 196-4 Recommended Terminology and Criteria for Neuroblastic Tumors | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Ultrastructurally, Taxy130 has described fine intracytoplasmic neurofilaments, dense-core neurosecretory granules, and abundant extracellular neurofibrillary material.
Clinical Features.
Mediastinal neuroblastomas are seen most often in children <1 year of age, but the tumor may occur in adolescents. Fifty percent are seen in children <1 year of age, and almost 90% are seen within the first 10 years of life.
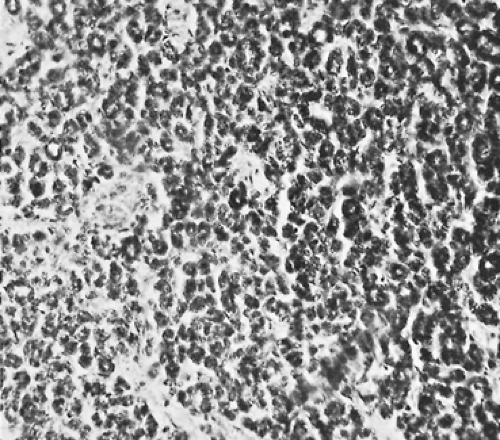 Figure 196-1. Photomicrograph of a neuroblastoma. Suggestion of pseudorosette formation is seen. |
These intrathoracic neuroblastomas account for approximately 15% to 20% of all neuroblastomas seen in the pediatric age group. Patients with mediastinal neuroblastomas may be asymptomatic and have only an incidental radiographic finding, but generally most patients are symptomatic with local and constitutional findings. Chest pain, Horner’s syndrome, paraplegia, cough, dyspnea, and dysphagia are not uncommon. Heterochromic iridis, fever, malaise, and failure to thrive can be present. As the result of production of catecholamines, sweating, flushing, or an admixture of both may develop. Diarrhea and abdominal distention may be observed and is probably due to the production vasoactive intestinal peptide. The more common complaints observed at the Children’s Memorial Hospital have been Horner’s syndrome, paraplegia, and flushing and sweating. Some infants also present with acute cerebellar ataxia with opsoclonus and chaotic nystagmus, the so-called dancing eyes originally described by Solomon and Chutorian122 and Altman and Baehner.5 Jones and associates58 believe that this syndrome is caused by an autoimmune mechanism. This symptom complex is seen in patients with more mature neuroblastomas and usually occurs in infants under 1 year of age. In 60% of infants with this syndrome, the neuroblastoma is located in the thorax, and most lesions are either stage 1 or 2 (Table 196-5). Stage 4 and 4S disease may occur in association with thoracic neuroblastomas, as with primary neuroblastomas elsewhere in the body. Distant metastases to liver or bone can occur, although thoracic neuroblastomas rarely present with metastatic involvement.
In addition to elevated plasma and urine levels of catecholamines, VMA, and HVA, elevated serum ferritin and lactate dehydrogenase levels may be present.
Radiographic Features.
Bar-Ziv and Nogrady11 describe the radiographic features of a paravertebral neuroblastoma as a ghost-like mass with indistinct borders (Fig. 196-2). These investigators described scattered calcifications in more
than 70% of these tumors, the uncalcified ones being more commonly seen in infants <1 year of age. Reed and associates101 found the incidence of calcification to be much less, but it is our impression that the incidence of calcification is greater than 50%. Rib erosions and rib displacement are common. A high incidence of intervertebral foramen enlargement and intraspinal canal extension is also present, occurring in two-thirds of the patients reported by Bar-Ziv and Nogrady.11 Reed and associates101 reported lower incidences of these bony changes.
than 70% of these tumors, the uncalcified ones being more commonly seen in infants <1 year of age. Reed and associates101 found the incidence of calcification to be much less, but it is our impression that the incidence of calcification is greater than 50%. Rib erosions and rib displacement are common. A high incidence of intervertebral foramen enlargement and intraspinal canal extension is also present, occurring in two-thirds of the patients reported by Bar-Ziv and Nogrady.11 Reed and associates101 reported lower incidences of these bony changes.
Table 196-5 Stage Description for Neuroblastomas | |||||||
|---|---|---|---|---|---|---|---|
|
Slovis and colleagues121 demonstrated that a chest radiograph is 100% sensitive in establishing a diagnosis of neuroblastoma. Magnetic resonance imaging (MRI) is the best imaging modality for detecting nodal involvement, intraspinal extension, and chest wall involvement (Fig. 196-3). A bone scan is needed to detect remote disease.
Staging.
The International Neuroblastoma Staging System (INSS) (Table 196-5) has replaced all other previously used staging systems and has been implemented worldwide.
Prognostic Factors.
Several clinical and biological prognostic factors have been identified in children with neuroblastoma (Table 196-6). Many of these prognostic factors play a role in treatment protocols. Stage of disease is the most important prognostic factor, and age at diagnosis is the only other independent prognostic indicator. Tumor markers predictive of poor outcome include serum ferritin (>142 ng/mL), serum lactic dehydrogenase (>1,500 IU/L), and serum neuron-specific enolase (>100 ng/mL).
Shimada and colleagues116 classified neuroblastoma histology as favorable or unfavorable depending on the degree of neuroblast differentiation, stromal content, mitosis–karyorrhexis index, and age at diagnosis (Table 196-7). Joshi and associates59 modified this system using mitotic ratio (i.e., number of mitoses per 10 high power fields) and the presence of calcification in the tumor (Table 196-8).
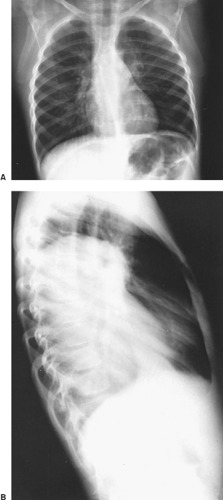 Figure 196-2. A,B: Posteroanterior and lateral radiographs of the chest of an infant with a large paraspinal neuroblastoma on the left. Note ghost-like shadow of the tumor on the posteroanterior view. (From Shields TW, Reynolds M. Neurogenic tumors of the thorax. Surg Clin North Am 1988;68:645. With permission.) |
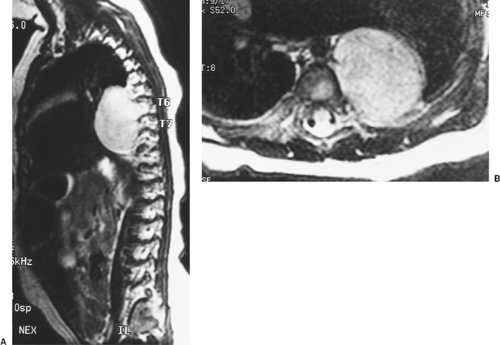 Figure 196-3. Lateral (A) and transverse (B) MRI scans demonstrate a thoracic neuroblastoma with intraspinal extension in this 4-month-old child. A combined thoracotomy and laminectomy was performed to remove the tumor. |
Amplification of the N-myc oncogene is associated with advanced-stage disease and rapid tumor progression, as described by Seeger112 and Brodeur17 and their associates. The relevance of N-myc amplification in low-stage or 4S disease is debated. Other biological prognostic factors include tumor cell ploidy; deletion or loss of heterozygosity of the short arm of chromosome 1; levels of expression of TrkA, TrkB, and TrkC (neutrophin receptors); levels of expression of multidrug resistance–associated protein; tumor vascularity; and expression of pCD44, a cell surface receptor.
In general, thoracic neuroblastomas have a favorable biological profile: low lactic dehydrogenase level, low serum ferritin level, DNA index = 1, and N-myc nonamplification. In the comprehensive review by Morris and colleagues,90 the biological variables measured for children with thoracic neuroblastoma did not completely explain the higher survival rates in this group when compared with the nonthoracic site (Figs. 196-4, 196-5, 196-6).
Treatment of Thoracic Neuroblastomas.
Clinical risk groups have been devised to classify patients with neuroblastoma and direct therapy. These risk groups are based on INSS stage, age, N-myc number, tumor cell ploidy, and tumor histology using Shimada criteria (Table 196-9).
Low-risk patients (INSS 1 and 2) and infants with 4S disease and favorable biology (normal N-myc copy number, hyperdiploid DNA index, and favorable histology) should be treated with complete surgical resection. Regional lymph nodes, especially the posterior mediastinal node groups, should be excised.
In patients with intraspinal extension, vertebral body involvement, or both, the cooperation of a neurosurgeon or an orthopedic spinal specialist is essential for evaluation and preoperative planning. Hoover and colleagues53 reported that recovery from neurologic deficits was not altered by laminectomy when chemotherapy or surgery was used. In their study comparing patients with and without laminectomy, there was a higher incidence of subsequent spinal deformities in those treated with laminectomy. Biopsy followed by chemotherapy may be the best preoperative strategy, as intraspinal extension may disappear and avoid the need for laminectomy.
Intermediate-risk patients (INSS 3 and 4) who lack N-myc amplification and infants with 4S tumor (normal N-myc copy number, diploid DNA content, or unfavorable histology) are treated with a combination of surgery and multiagent chemotherapy. Few patients with thoracic neuroblastoma fit into this risk group.
The indications for and timing of surgery for patients in the high-risk group are controversial. Initial surgical resection of large tumors is seldom recommended. Multimodal therapies include myeloablative chemotherapy or chemoradiation therapy followed by autologous bone marrow transplantation. Kletzel and associates67 reported improved survival statistics for patients with high-risk neuroblastoma.
Thoracoscopy is being used with increasing frequency for diagnostic biopsy and complete surgical resection of thoracic
neurogenic tumors. Petty and colleagues99 compared open thoracotomy with thoracoscopy in a small group of patients and found similar results in terms of local control and disease-free interval. Thoracoscopy allowed shorter length of hospital stay. DeCou and associates30 reported excellent results in a small series of patients who underwent primary gross total resection with the thoracoscopic approach. All were stage I and most had favorable histology. None were N-myc–amplified.
neurogenic tumors. Petty and colleagues99 compared open thoracotomy with thoracoscopy in a small group of patients and found similar results in terms of local control and disease-free interval. Thoracoscopy allowed shorter length of hospital stay. DeCou and associates30 reported excellent results in a small series of patients who underwent primary gross total resection with the thoracoscopic approach. All were stage I and most had favorable histology. None were N-myc–amplified.
Table 196-6 Prognostic Factors in Neuroblastomas Analyzed Singly | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ganglioneuroblastoma
Ganglioneuroblastoma is a malignant tumor but less aggressive than neuroblastoma. Grossly, the tumor is firm in consistency in two-thirds of cases. Adam and Hochholzer2 reported that of the 48 lesions in which encapsulation was recorded, 69% were grossly encapsulated, 27% were partially encapsulated, and only 4% were without a capsule. Microscopically, ganglioneuroblastomas are composed of a mixture of mature ganglion cells and neuroblasts set in a pink fibrillar background (Fig. 196-7).
Table 196-7 Shimada Histologic Grading System for Neuroblastoma | ||
|---|---|---|
|
Table 196-8 Association of Favorable Histologic Features With Prognostic Subgroups of the Shimada Classification | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Overall, these tumors are less common than neuroblastomas, but in the thorax they appear to have approximately the same or a slightly greater incidence of occurrence than the neuro- blastomas. In Adam and Hochholzer’s2 review of the autonomic neurogenic lesions in the paravertebral sulci in the files of the AFIP, there were 65 neuroblastomas and 80 ganglioneuroblastomas. These lesions are reported to be seen more often in older children and adolescents than are the neuroblastomas (see Table 196-3). However, in Adam and Hochholzer’s series, one-third of the patients were ≤2 years of age, half were ≤3 years of age, and four-fifths were ≤10 years of age. Only 10 cases were seen between the ages of 12 and 20 years, and only 3 patients were >20 years of age. Boys and girls <10 years of age were affected equally, although a slight predominance was seen for girls aged >12.
Approximately half of these tumors are discovered as an asymptomatic mass on routine radiography of the chest. The other patients present with findings similar to those noted for neuroblastoma, although clinical evidence of excessive catecholamines is only infrequently noted and laboratory evidence of elevated VMA or HVA is found in only 12% of patients. Evidence of extension into the spinal canal is likewise uncommon.
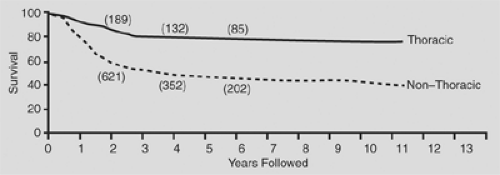 Figure 196-4. Survival curves for thoracic (n = 227) and nonthoracic patients (n = 1,108) (p < 0.0001). Numbers above the curves represent cases with follow-up up to or beyond the specified time points. (From Morris JA, et al. Biological variables in thoracic neuroblastoma: a Pediatric Oncology Group Study. J Pediatr Surg 1995;30:296. With permission.) |
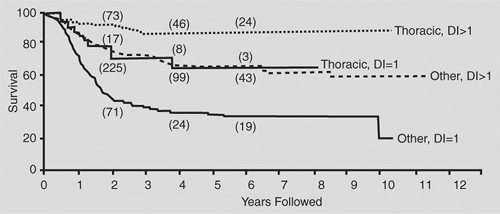 Figure 196-5. Survival curves for thoracic (n = 114) and nonthoracic (n = 540) patients for whom the DNA index was recorded (stratified log-rank test; p < 0.0001). Numbers above the curves represent cases with follow-up up to or beyond the specified time points. (From Morris JA, et al. Biological variables in thoracic neuroblastoma: a Pediatric Oncology Group Study. J Pediatr Surg 1995;30:296. With permission.) |
The radiographic features of the lobulated or oval paraspinal mass tend to be more distinct than the ghost-like shadow of the neuroblastoma (Fig. 196-8). Stippled calcification may be present. Chest wall changes (i.e., rib erosion or displacement according to Reed and associates101) occur in only 5% to 10% of patients. Extension into the spinal canal is, as noted, uncommon, but MRI is indicated in all cases to rule it out.
Most children with ganglioneuroblastoma present with a solitary mass that can be resected. Complete evaluation of the patient and tumor analysis for biologic variables are critical to identify the more aggressive tumors. INSS staging and clinical risk-group assignment should be made. Treatment is primarily surgical, although chemotherapy is indicated in the intermediate- and high-risk groups.
Ganglioneuromas
Ganglioneuromas are benign tumors and represent the full maturation of autonomic nervous tissue tumors. They may arise de novo or may follow maturation of a neuroblastoma. They account for approximately 42% of the autonomic neural crest tumors.
Pathology.
Ganglioneuromas are frequently large, firm, well-circumscribed, encapsulated tumors arising in a paravertebral sulcus in a child, adolescent, or adult. The tumor is usually attached to a sympathetic or intercostal nerve trunk. Extension
into the spinal canal can occur but is uncommon. On cut section, the tumor is yellowish to gray; Marchevsky and Kaneko79 state that it frequently has a whorled, trabeculated surface that may simulate a leiomyoma. Microscopic examination reveals mature ganglion cells, singly and in nests, in a stroma composed of Schwann cells and fibrous tissue (Fig. 196-9). Areas of degeneration are evident within the tumor. Calcification may or may not be present in these areas. The ganglion cells are large, and their cytoplasm may contain various inclusions. Ultrastructurally, dense-core vesicles have been identified by Bender and Ghatak13 and Yokoyama and associates142 as well as others.
into the spinal canal can occur but is uncommon. On cut section, the tumor is yellowish to gray; Marchevsky and Kaneko79 state that it frequently has a whorled, trabeculated surface that may simulate a leiomyoma. Microscopic examination reveals mature ganglion cells, singly and in nests, in a stroma composed of Schwann cells and fibrous tissue (Fig. 196-9). Areas of degeneration are evident within the tumor. Calcification may or may not be present in these areas. The ganglion cells are large, and their cytoplasm may contain various inclusions. Ultrastructurally, dense-core vesicles have been identified by Bender and Ghatak13 and Yokoyama and associates142 as well as others.
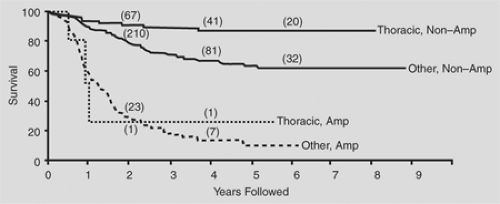 Figure 196-6. Survival curves for thoracic (n = 84) and nonthoracic (n = 409) patients for whom N-myc amplification was recorded (stratified log-rank test; p = 0.003). Numbers above the curves represent cases with follow-up up to or beyond the specified time points. (From Morris JA, et al. Biological variables in thoracic neuroblastoma: a Pediatric Oncology, Group Study. J Pediatr Surg 1995;30:296. With permission.) |
Table 196-9 Risk Groups for Neuroblastoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
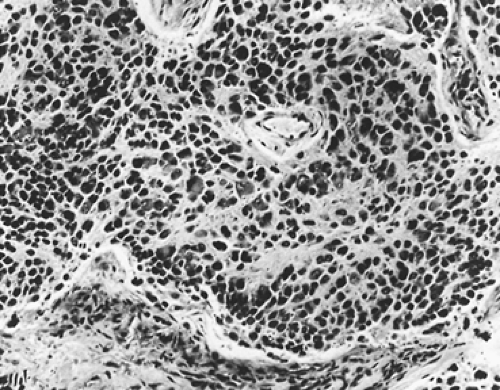 Figure 196-7. Ganglioneuroblastoma composed of round neuro- blastic cells and larger ganglion cells. (From Marchevsky AM, Kaneko M. Surgical Pathology of the Mediastinum. New York: Raven Press, 1984. With permission.) |
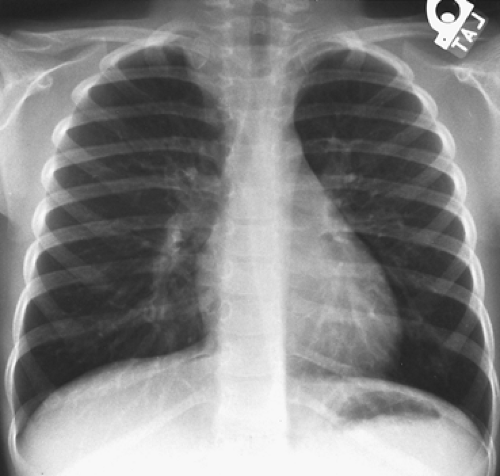 Figure 196-8. Chest radiograph of a mass in the apical portion of the right paravertebral sulcus. The radiographic shadow is much more distinct than that of a neuroblastoma. On surgical excision, the tumor proved to be a ganglioneuroblastoma. |
Clinical Features.
Clinically, the patient may be asymptomatic, but patients with large lesions may present with cough, dyspnea, dysphagia, Horner’s syndrome, and, occasionally, chest pain. Heterochromia iridis was reported by McRae
and Shaw83 to be the result of sympathetic injury by the tumor involving the superior cervical ganglion. The age distribution is not dissimilar to that observed in patients with ganglioneuro- blastomas (see Table 196-3). Elevated levels of VMA and HVA are observed infrequently. Chronic diarrhea also has been reported in association with these tumors by Mendelsohn84 and Trump133 and their associates. This is thought to be caused by the presence of vasoactive intestinal peptide, which can be localized to the cytoplasm of the ganglion cells by means of immunoperoxidase techniques.
and Shaw83 to be the result of sympathetic injury by the tumor involving the superior cervical ganglion. The age distribution is not dissimilar to that observed in patients with ganglioneuro- blastomas (see Table 196-3). Elevated levels of VMA and HVA are observed infrequently. Chronic diarrhea also has been reported in association with these tumors by Mendelsohn84 and Trump133 and their associates. This is thought to be caused by the presence of vasoactive intestinal peptide, which can be localized to the cytoplasm of the ganglion cells by means of immunoperoxidase techniques.
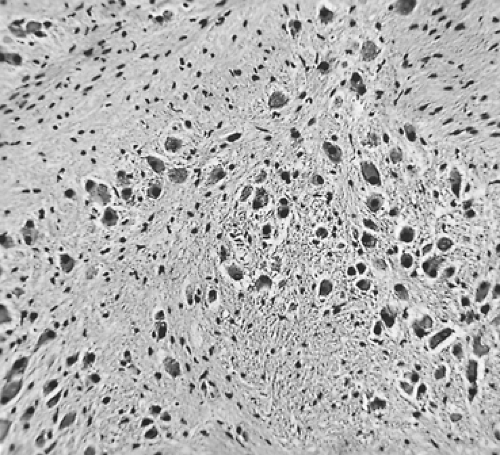 Figure 196-9. Photomicrograph of a typical ganglioneuroma. |
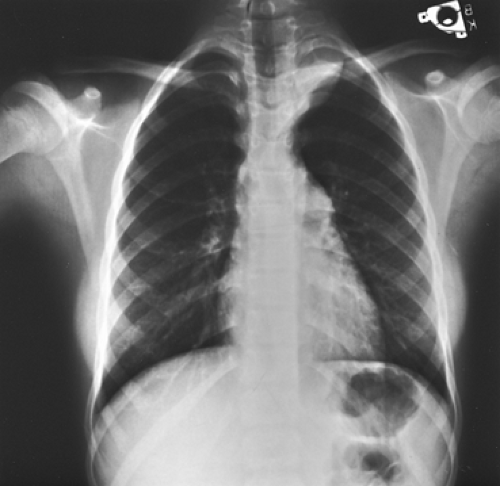 Figure 196-10. Posteroanterior radiograph of the chest of an adolescent girl with an asymptomatic left superior mediastinal mass. On histologic examination after surgical excision, the lesion proved to be a ganglioneuroma. |
Radiographic Features.
The tumor presents as a solid, well-defined, ovoid or lobulated mass in one of the paravertebral sulci (Fig. 196-10). Stippled calcification was observed by Bar-Ziv and Nogrady11 in 50% of the patients studied. Rib erosion, when observed, is uncommon and subtle in nature. Extension into the spinal canal is uncommon but can occur, as noted by Bar-Ziv and Nogrady11 and Davidson and associates.28 Thus, computed tomographic (CT) or MRI scanning is always indicated (Fig. 196-11).
Treatment.
Surgical excision is the treatment of choice and is curative. Marchevsky and Kaneko78 report that occasionally a regional lymph node may contain metastatic ganglioneuroma. Enzinger and Weiss35 believe these to be residual foci of neuroblastomas that have matured into benign ganglioneuromas; subsequent recurrence of the disease is not seen in these patients.
Tumors of Nerve Sheath Origin
Benign Nerve Sheath Tumors
Both schwannomas (neurilemomas) and neurofibromas may occur in patients <20 years of age. In the series reported by Reed and associates,101 15% of the neurogenic intrathoracic tumors seen in these age groups were of nerve sheath origin (see Table 196-3). These tumors are more common in patients between the ages of 16 and 20 years, although two cases of schwannomas were seen in children <10 years of age; five cases of nerve sheath origin were seen in this younger age group in Ribet and Cardot’s103 series. The incidence of the two varieties is approximately equal, but the neurofibromas are more often seen in patients with von Recklinghausen’s disease. Incidentally, neuroblastomas are also often observed in patients with this disease.
These benign nerve sheath tumors are found almost exclusively in the paravertebral sulci. Their pathologic and clinical features are the same as those of the adult population. These features—as well as their diagnostic investigation, treatment, and prognosis—are discussed in detail subsequently in this chapter.
Malignant Nerve Sheath Tumors
Malignant schwannoma (neurogenic sarcoma) rarely occurs in childhood. Isolated mention of its occurrence in series of
childhood neurogenic thoracic tumors has been recorded; one such patient was seen at the Children’s Memorial Hospital in Chicago. This lesion’s features are not dissimilar from those observed in adults; these features are discussed later in this chapter. It may be noted that Enzinger and Weiss35 as well as Keller63 and Ricci104 and their associates have described rare instances of malignant transformation of ganglioneuromas into malignant schwannomas. Also, malignant transformation of benign neurofibromas has been described, especially in children with von Recklinghausen’s disease. Long-term observation over 10 to 20 years may reveal the occurrence of this event. A rapid increase in size of a benign lesion or the development of pain is indicative of malignant change.
childhood neurogenic thoracic tumors has been recorded; one such patient was seen at the Children’s Memorial Hospital in Chicago. This lesion’s features are not dissimilar from those observed in adults; these features are discussed later in this chapter. It may be noted that Enzinger and Weiss35 as well as Keller63 and Ricci104 and their associates have described rare instances of malignant transformation of ganglioneuromas into malignant schwannomas. Also, malignant transformation of benign neurofibromas has been described, especially in children with von Recklinghausen’s disease. Long-term observation over 10 to 20 years may reveal the occurrence of this event. A rapid increase in size of a benign lesion or the development of pain is indicative of malignant change.
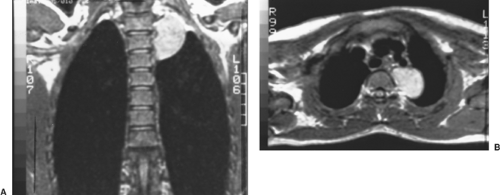 Figure 196-11. A, B: Coronal and transverse MRI scans of the mass shown in Figure 196-8 revealed no extension into the spinal canal. |
Tumors of Neuroectodermal Origin
Two tumors of presumed neuroectodermal origin may occur in the mediastinum. One is the pigmented neuroectodermal tumor of infancy (melanotic progonoma) and the second is the malignant small cell tumor of the thoracopulmonary region (Askin’s tumor) seen in the older child or adolescent.
Melanotic Progonoma
Melanotic progonoma is seen most commonly in the upper or lower jaws but infrequently may occur in the mediastinum where it can be confused with a pigmented neuroblastoma.
Marchevsky and Kaneko79 describe these tumors as dark gray or black tumors that histologically are composed of irregular spaces lined by cuboidal cells containing intracytoplasmic melanin and ultrastructural features of epithelium and melanocytes. Misugi and associates87 and Williams138 considered these to be malignant tumors, which recur in 15% of cases and may metastasize widely. Treatment of this unusual tumor is local excision. The efficacy of other treatment modalities is unknown.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


