Benign and Malignant Neoplasms of the Mediastinum
INTRODUCTION
Primary lesions of the mediastinum are less common than lesions that secondarily involve the mediastinum. Overall, the majority of masses discovered in the mediastinum will be found to be metastases from a primary lung cancer. Neoplasms that arise primarily in the mediastinum, however, are often encountered in the clinic and are represented by a variety of lesions. This chapter first reviews the anatomy of the mediastinum and then focuses on the benign and malignant neoplasms that arise within each of the anatomic regions of the mediastinum. Covered elsewhere in this textbook (see Chapter 80) are nonneoplastic disorders of the mediastinum that include pneumomediastinum, acute mediastinitis, chronic mediastinitis, and other miscellaneous disorders. Likewise, congenital lesions of the mediastinum, such as bronchogenic, enterogenous, neurogenic, thymic, pericardial, and thoracic duct cysts, are also covered elsewhere (see Chapter 81).
ANATOMY OF THE MEDIASTINUM
The mediastinum comprises an anatomic space located between the thoracic inlet and the diaphragm, and bordered on the left and right sides by the pleural cavities. This central anatomic location houses or borders vital structures of almost every major organ system including the heart and great vessels of the circulatory system, the esophagus, and major airways of the aerodigestive tract, the thymus of the immune system, and important nerves such as the phrenic and vagus nerves. Further, various endocrine organs may project into it, distant malignancies may metastasize to it, and infectious processes can manifest themselves within it.
The mediastinum is compartmentalized based upon the borders of anatomic structures as seen on a lateral chest radiograph (Fig. 82-1). We believe that the most anatomically appropriate and clinically useful model of the mediastinum is the three-compartment model.1 The three-compartment scheme divides the mediastinum into anterior, middle, and posterior compartments. Here, the anterior mediastinum extends from the thoracic inlet superiorly to the diaphragm inferiorly and is bounded anteriorly by the posterior table of the sternum, and posteriorly by the anterior pericardium and the aorta, innominate vein, and brachiocephalic vessels. The content of the anterior compartment includes the thymus, variable amounts of fat and lymphatic tissues, and the internal mammary arteries and veins. The middle compartment of the mediastinum is bounded anteriorly by the pericardium and posteriorly by the pericardium and posterior wall of the trachea, extending only as high as the pericardial reflection. The middle mediastinum contains the heart, pericardium, superior and inferior vena cava, ascending and transverse aorta, trachea and mainstem bronchi, and lymphatic tissues. The posterior mediastinum extends from the thoracic inlet to the diaphragm, and in this “three-compartment scheme,” lies posterior to the posterior pericardium and airway. It includes the descending aorta, thoracic duct, esophagus, vagus nerves, and lymph nodes, as well as structures emerging from the spinal canal such as intercostal nerves.
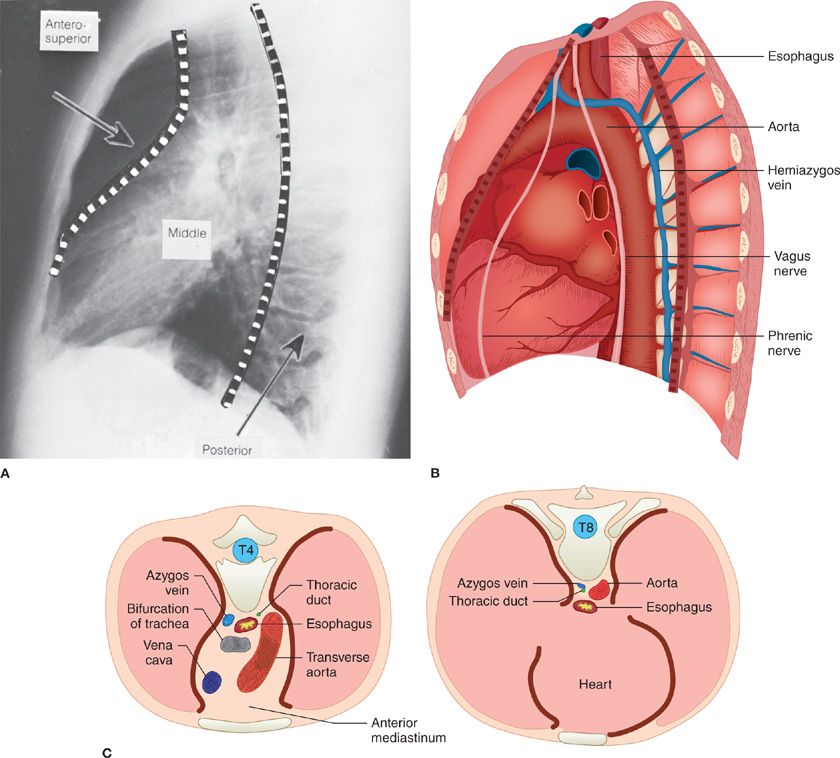
Figure 82-1 Three-compartment model of the mediastinum. A. Lateral radiograph of the chest. B. Schematic representation of the contents of the three compartments of the mediastinum. C. Cross sections of the thorax at T4 (left) and T8 (right) to show relative positions of mediastinal structures.
One of the main advantages of categorizing the anatomy of the mediastinum is the ability it confers to generate a differential diagnosis for a mediastinal mass based on the structures naturally contained within the compartment in which it arises (Table 82-1). Such a classification scheme allows a clinician to proceed systematically to determine the appropriate diagnostic or therapeutic approach depending on the differential diagnosis.
EPIDEMIOLOGY AND INCIDENCE
The incidence and type of primary mediastinal neoplasms varies with patient age. In combined series totaling 3017 mostly adult patients, the incidence of mediastinal masses in decreasing frequency were thymomas and thymic cysts (26.5%), neurogenic tumors (20.2%), germ cell tumors (GCTs) (13.8%), lymphomas (12.7%), foregut cysts (10.3%), and pleuropericardial cysts (6.6%). In children, combined series totaling 718 patients demonstrated that neurogenic tumors were most common (41.6%), followed by GCTs (13.5%), foregut cysts (13.4%), lymphomas (13.4%), angiomas and lymphangiomas (6.1%), and thymic tumors or cysts (4.9%).2 In general, the incidence of anterior lesions is higher in adults, and posterior lesions predominate in children. Further, the incidence of malignancy differs among primary mediastinal masses arising in each of the different compartments. In one of the largest series, Davis and colleagues demonstrated that among patients with mediastinal masses, malignancy was found in 59% of those in the anterior mediastinum, 29% of those in the middle mediastinum, and 16% of those in the posterior mediastinum.3
As discussed below, mediastinal masses are often incidentally detected on imaging studies obtained for other reasons. An estimate of the frequency of “incidental” mediastinal masses has been provided by a large lung cancer screening study. In 9263 individuals at high risk for lung cancer who underwent a computed tomography (CT) screening examination, a mediastinal mass was found in 71 patients (0.77%). The majority of these incidental masses were thymic and most were treated successfully with a conservative approach.4
SIGNS AND SYMPTOMS
Signs and symptoms of mediastinal tumors can vary widely at presentation and usually offer only nonspecific clues to the nature of the underlying disease process. While over 60% of patients do present with symptoms, it is also quite common for an asymptomatic mass to be detected on a routine screening examination. Most asymptomatic patients with a mediastinal mass will have a benign lesion, while the majority of patients that present with symptoms have an underlying malignant process. In Davis’ series of 400 patients with mediastinal masses, 83% of the lesions found on routine chest radiographs were benign, whereas 57% of lesions in symptomatic patients were malignant.3
The presenting symptoms of a mediastinal mass vary widely and are influenced by anatomic location and the presence of malignant invasion or mass effect. Dyspnea or cough may result from airway invasion or abutment, tamponade, or the presence of a pleural effusion. Dysphagia is seen with esophageal compression, and chest pain may represent chest wall or neural invasion or abutment. Invasion of the airway may result in hemoptysis, invasion of the recurrent laryngeal nerve may present as hoarseness, and invasion of the superior vena cava can present with facial swelling and superior vena cava syndrome. Constitutional symptoms such as fever and night sweats are often associated with mediastinal lymphoma, and myasthenic symptoms may be suggestive of thymoma.
ANTERIOR MEDIASTINAL NEOPLASMS
Neoplasms of the anterior mediastinum are discussed below.
 EVALUATION OF THE ANTERIOR MEDIASTINAL MASS
EVALUATION OF THE ANTERIOR MEDIASTINAL MASS
In a patient with an anterior mediastinal mass, it is often possible to make a strong provisional diagnosis based upon clinical evaluation and imaging data.5 Whereas specific workup for common anterior mediastinal mass lesions is described in the following sections, some general principles are reviewed here.
History and Examination
In an individual younger than 40 years, lymphoma is the most likely diagnosis and the presence of International Working Formulation (IWF) group B symptoms or palpable lymphadenopathy further increases the level of suspicion. In contrast to lymphoma, thymic neoplasms are very uncommon before the fourth decade of life. The presence of a paraneoplastic syndrome associated with an anterior mediastinal mass essentially clinches the diagnosis of thymoma. The majority of GCTs (benign or malignant) are diagnosed in the second or third decade of life. Whereas patients with a thymoma often have an indolent presentation, patients with a lymphoma or a malignant GCT often have a rapid onset of symptoms. Workup must include examination of the testes to rule out a testicular primary GCT.
Serum Studies
Autoantibodies to the acetylcholine receptor (anti-AChR) should be measured as their presence is virtually diagnostic of myasthenia gravis, even if the patient is without obvious symptoms. Characteristic serum tumor markers such as beta-human chorionic gonadotropin (β-HCG) and alpha-fetoprotein (AFP) are elaborated by most malignant GCTs but not by benign GCTs.6 Elevated serum LDH is suggestive of lymphoma. Evidence of hypo- or hyperthyroidism, as measured by thyroid-stimulating hormone (TSH) and thyroid hormones (T3 and T4), suggests mediastinal goiter, though goiter is not invariably associated with abnormal hormone production.
Imaging Studies
CT provides valuable data about the anatomic location of the tumor, its physical characteristics (fatty, solid, or cystic) and degree of invasiveness. Occasionally, magnetic resonance imaging (MRI) provides useful additional information concerning obliteration of normal tissue planes and vascular invasion. Although imaging is most often not diagnostic of the specific type of tumor, some imaging features can be pathognomonic. For example, the finding of a well-encapsulated lesion in the anterior mediastinum containing several tissue elements – calcium, fat, fluid – is essentially diagnostic of a mature teratoma.
Need for Biopsy
The decision whether to perform a biopsy of an anterior mediastinal mass is not simple. Routine biopsy should not be endorsed, not only because of the unnecessary morbidity and costs associated with the procedure, but also because of the potential risk of tumor spread. Well-encapsulated lesions believed not to be lymphoma are often resected for both diagnosis and treatment, without a preceding biopsy. Conversely, for locally invasive or frankly unresectable anterior mediastinal masses, a biopsy should often be performed as such lesions may represent lymphoma, aggressive thymomas that could benefit from neoadjuvant treatment, malignant GCTs, or other rare diseases.
Once a clinical diagnosis of thymoma is made, the goal is to proceed directly to resection without preliminary biopsy, as these tumors have a predilection for local recurrence once the thymic capsule has been violated. When palpable peripheral nodes are present, the diagnosis of lymphoma is often most easily obtained by excision of one of them. Patients with suspected lymphoma and an isolated anterior mediastinal mass should undergo either a CT-guided core needle biopsy or a Chamberlain procedure (anterior mediastinotomy) for diagnosis, depending upon the pathologist’s level of comfort in classifying lymphoma on the basis of specimen size at one’s institution. Resection of lymphoma is not indicated and can almost always be avoided by performing a diagnostic biopsy when lymphoma is suspected. Surgical extirpation is the mainstay of treatment for mature GCTs, and biopsy is not indicated for these lesions if they have the characteristic imaging characteristics. Malignant GCTs, on the other hand, are treated primarily with chemotherapy, radiotherapy, or both. In situations where these lesions are suspected but tumor markers (AFP and β-HCG) are not markedly elevated, biopsy should be performed.
 THYMOMA
THYMOMA
Clinical assessment and management of thymoma are discussed below.
Presentation
The age distribution of patients with a thymoma is represented by a broad peak between approximately 35 and 70 years with a median age of about 54 years.7 The ratio of men to women with this disease is about equal. Approximately one-third of patients with a thymoma will not report significant symptoms and an additional one-third will report symptoms of cough, dyspnea, or chest pain reflective of compression or invasion of adjacent structures. Patients may also present with symptoms of an associated paraneoplastic syndrome. Forty to 45% of patients with a thymoma will present with myasthenia gravis and, conversely, 5% to 15% of patients with myasthenia gravis will be found to have a thymoma.8,9 Notably, myasthenia gravis can develop some time after diagnosis of thymoma, and occasionally following resection, highlighting the necessity of complete resection of both the thymoma and the entire thymus gland. Thymomas may also be associated with pure red cell aplasia, agammaglobulinemia, systemic lupus erythematosus, and other autoimmune disorders.
Diagnosis
A clinical diagnosis of thymoma is often sufficient to proceed to resection without biopsy. The presence of a paraneoplastic syndrome associated with an anterior mediastinal mass essentially clinches the diagnosis of thymoma. Autoantibodies to the acetylcholine (Ach) receptor should be measured, and their presence is virtually diagnostic of myasthenia gravis and also clinches the diagnosis of thymoma. Interestingly, Ach receptor antibodies are demonstrated in approximately 60% of patients who have thymoma without neurologic symptoms.10 It is not necessary to obtain a preoperative tissue biopsy for a small, resectable anterior mediastinal tumor whose radiographic features are typical of thymoma in an asymptomatic patient. Although there are no pathognomonic imaging features that differentiate thymoma from several other mediastinal tumors, in a patient without B-symptoms to suggest lymphoma, the diagnosis of thymoma is strongly suggested by a CT scan demonstrating a well-circumscribed, solid anterior mediastinal mass without the low-density areas that would suggest the cystic and fatty components of a teratoma (Fig. 82-2). Most thymomas are solid tumors, but up to a third may have components that are necrotic, hemorrhagic, or cystic. Several small studies have investigated the utility of PET in evaluation of thymoma and have suggested that FDG uptake is greater in thymoma than in thymic hyperplasia, and that thymic carcinomas demonstrate the highest FDG avidity.11 However, PET is not required to stage a thymoma preoperatively when it appears noninvasive/nonmalignant on CT.
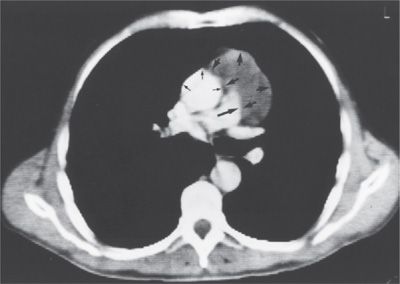
Figure 82-2 Thymoma. Axial section from a CT scan demonstrates a 4-cm mass abutting the thoracic aorta without any evidence of invasion (arrows).
Once a diagnosis of thymoma is suggested, the goal is to proceed directly to resection without preliminary biopsy, as these tumors have a predilection for local recurrence once the capsule has been violated. A definitive tissue diagnosis is needed primarily when a presumed thymoma is so advanced that it would be best treated either nonoperatively or with neoadjuvant chemotherapy or chemoradiotherapy, or in instances where there is a strong possibility of lymphoma. If required, the pathologic diagnosis of thymoma can usually be achieved by image-guided core needle biopsy, or if that fails, through open surgical biopsy. The success rate of needle biopsy in establishing the diagnosis of thymoma is approximately 60% and the success rate of surgical biopsy is approximately 90%.12 In our opinion, video-assisted thoracoscopic surgical (VATS) biopsy of an anterior mediastinal mass in which thymoma is in the differential should be assiduously avoided, due to the great potential of spread into the pleural space. If core needle biopsy fails and a diagnosis is required, a Chamberlain procedure (anterior mediastinotomy) should be performed.
All patients with suspected thymoma should be evaluated for myasthenia gravis. This evaluation usually begins with a careful assessment for the presence of ocular, bulbar, and limb muscle weakness. The diagnosis of myasthenia gravis in a patient with a characteristic history and physical examination requires two positive confirmatory tests among pharmacologic (Tensilon test), serologic (anti-AChR antibodies), and electrodiagnostic (EMG) studies. If there is any suggestion of myasthenia gravis on initial presentation, the patient should undergo preoperative evaluation by a neurologist. Medical optimization prior to surgery using some combination of cholinesterase inhibitors, intravenous immunoglobulin, plasmapheresis, and occasionally steroids, can help avoid respiratory failure in the perioperative period. Other paraneoplastic syndromes are also associated with thymoma, including hypogammaglobulinemia in 10% of patients, and pure red cell aplasia in 5% of patients.
Staging
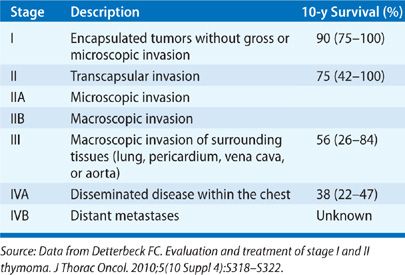
The most widely utilized classification scheme for thymoma was proposed by Masaoka in 1981 and is based on the local invasiveness of the tumor (Table 82-2).13 The Masaoka classification system has prognostic value14 and permits stratification for adjuvant therapy. One difficulty with this system is that it is based upon the findings at the time of surgical resection, thus precluding its use for triaging some patients to neoadjuvant therapy.
Thymomas are also classified histologically. A thymoma is composed of a mixture of thymic epithelial cells with bland features as well as lymphocytes in various stages of development. The neoplastic cell of origin is believed to be the thymic epithelial cell and not the lymphocyte,15 and thymomas appear histologically benign even when they are invasive. In 1985, Müller-Hermelink proposed a novel histologic classification system to separate all thymomas into categories of cortical, medullary, or mixed, based on the histologic appearance of the thymic epithelial cells.16 Cortical thymomas are composed of large, round, or polygonal epithelial cells and medullary thymomas contain smaller, spindle cell–shaped epithelial cells with irregular nuclei. This histologic classification system was found to reliably predict tumor behavior and prognosis, with medullary thymomas behaving in an essentially benign fashion and cortical thymomas more frequently demonstrating evidence of invasive and malignant disease.17 Later studies have demonstrated that both the Masaoka staging and Müller-Hermelink grading systems are strong and independent prognostic indicators of both overall as well as disease-free survival.14,18,19
More recently, the World Health Organization (WHO) proposed a histologic classification system (resembling the Müller-Hermelink system) that has become the histologic grading system of choice in the current era and is useful to distinguish between thymoma, thymic carcinoma, and thymic carcinoids (Table 82-3).20 It is now accepted that the Masaoka staging criteria used in combination with the WHO grading criteria provide the most accurate prognostic information for survival and recurrence in thymoma.21,22

Treatment
Surgical resection is the mainstay of treatment of Masaoka stage I–III thymoma while the treatment of stage IVa thymoma is controversial. Resection of even a small, stage I thymoma without myasthenia gravis should include removal of the entire thymus en bloc along with the tumor because (1) myasthenia gravis can potentially develop postoperatively (and total thymectomy is an appropriate treatment for myasthenia gravis), and (2) a second focus of tumor within the thymus is occasionally found. The gold standard surgical approach to thymoma remains median sternotomy but minimally invasive approaches including VATS and robotic techniques are likely equally effective for smaller tumors in experienced hands. Certainly if there is suspicion of invasion into adjacent structures, sternotomy is the preferred approach as it is critical that the involved portions of these structures are resected en bloc with the thymus gland and tumor. This can require resection of lung, pericardium, innominate vein, superior vena cava, phrenic nerve, and even aorta.
Stage I thymomas are treated adequately with complete resection alone. Although in the past adjuvant radiotherapy was frequently administered to patients with stage II thymoma, accumulated data suggest that adjuvant radiotherapy is of little additional benefit following complete resection.23–25 Thymoma is a radiosensitive tumor however, and for patients who have incomplete resection, postoperative radiotherapy is recommended. For patients with advanced disease (stage III or IV thymoma), chemotherapy is recommended and is often followed by radiation for patients with incompletely resected disease. For thymoma that is initially considered unresectable, induction chemotherapy followed by surgical resection can result in favorable rates of overall and disease-free survival.26,27 Further, surgical resection plays an important role in the management of recurrences and complete resection of a recurrence can result in improved overall survival.28
Outcome
Although thymomas are generally indolent tumors, they should be considered a malignant neoplasm as they have the ability to metastasize to the pleura, pericardium, and (less commonly) distant sites. One study reported that 4% of 207 patients evaluated for surgical resection of a thymoma had lymph node or distant metastases at presentation, and another 16% developed nodal or distant metastases at some point in their course.29 Others have shown that distant metastases can occur with any stage or histology.30,31 Detterbeck reviewed overall survival in compiled surgical series of at least 100 patients (2437 patients total) and demonstrated favorable 10-year survival rates of approximately 90% and 70% for stage I and II, and 55% and 35% for stage III and IVa thymoma, respectively.12 Completeness of resection is the most important predictor of recurrence and survival; significantly better survival is demonstrated when resection is complete in all large studies examining this issue.
Locoregional recurrence is far more common than distant recurrence, and about half of all local recurrences involve the pleural space or the lung. The average recurrence rate for stage I tumors is 3% but increases to 11% and 30% for stage II and III tumors, respectively.7 The relatively indolent nature of a thymoma is reflected by an average time to recurrence of 5 years, with rare recurrences seen as far out as 20 years. The presence of myasthenia gravis is no longer considered a negative prognostic factor, as it was in older studies, and over 30% of patients with myasthenia and thymoma ultimately achieve a complete remission of the myasthenia following thymectomy.32 Similarly, approximately one-third of patients with associated red cell aplasia experience improvement following thymectomy. In contrast, hypogammaglobulinemia associated with thymoma generally does not respond to thymectomy.
 THYMIC CARCINOMA
THYMIC CARCINOMA
Clinical assessment and management of thymic carcinoma are discussed below.
Presentation
Thymic carcinomas (WHO type C) are highly aggressive neoplasms of thymic epithelial origin and are very different from thymomas (WHO types A, AB, and B). Thymic carcinomas are rare, constituting approximately 10% of all thymic neoplasms.28 They can occur at any age but are most frequently observed in persons between 30 and 60 years of age. The majority of patients with thymic carcinoma present with symptoms of local invasion or compression such as cough, chest pain, or superior vena cava syndrome. Pericardial and/or pleural effusions are often seen. Thymic carcinoma is typically not associated with myasthenia gravis. Unlike thymomas, thymic carcinomas frequently metastasize to lymph nodes and distant sites. Eighty percent of patients have local invasion of contiguous mediastinal structures at the time of presentation, and 40% of cases have metastatic spread to bones, lung, pleura, liver, or lymph nodes.33 Over half of the patients will present with locally advanced, although potentially resectable, disease.34,35
Diagnosis
Thymic carcinomas can be distinguished from thymomas based on their malignant histologic features and different immunohistochemical and genetic characteristics. Imaging studies often reveal an invasive presentation, and for this reason, percutaneous needle biopsy should generally be undertaken.
Staging
The Masaoka staging system and WHO histologic classification system are used to stage thymic carcinoma. In the WHO system, thymic carcinomas are type C lesions. These tumors are distinct from thymoma and should not be considered in the same light as a thymoma with local invasion. Histologically, thymic carcinoma contains a number of different cell types, but they are unified by their unequivocal malignant appearance on light microscopy. Division of patients into those with low-grade histology and those with high-grade histology has prognostic significance. The median survival for patients with low-grade histology (squamous, mucoepidermoid, and basiloid thymic carcinomas) is 29 months, as compared to 11 months for patients with high-grade histology (sarcomatoid and clear cell thymic carcinoma).36
Treatment
While there is no standard-of-care approach to patients with thymic carcinoma, a multidisciplinary strategy is recommended. The prevailing practice is that patients with stage I–III and some IVa patients should be treated with some combination of surgical resection plus chemotherapy and/or radiotherapy.37 Although thymic carcinomas generally respond poorly to chemotherapy, carboplatin and paclitaxel are recommended because this combination has shown the highest response rates in clinical trials. For the rare thymic carcinoma patients with clearly resectable disease, surgery is considered the primary therapeutic modality. For those with disease thought to be initially unresectable or disease that appears clearly to be invading one or more surrounding organs or major vascular structures, neoadjuvant chemotherapy and/or radiation may improve operability and permit subsequent resection.36,38,39
Outcome
The prognosis for patients with thymic carcinoma is much worse than for patients with thymoma, with 5-year overall survival rates for thymic carcinomas in the range of 30% to 50%.35,40,41 A more recent clinicopathologic study of 65 cases of primary thymic carcinoma, however, reported a more favorable 5-year survival rate of 66%. In this study, 63% of the patients received additional therapy in the form of chemotherapy or radiation.34 Similar to thymoma, patients with completely resected thymic carcinomas have longer overall and progression-free survival rates than those who undergo incomplete resection or are unresectable. In a recent series of 60 patients with thymic carcinoma (40 treated surgically), completely resected lesions and early Masaoka stages are the most important factors associated with successful disease control and long-term survival. Five-year survival of 85% was obtained after complete resection, compared with 29% in those with incomplete resection.42 In another series of 16 patients, a multimodality approach that included surgery resulted in complete resection in 88% of the patients and a mean survival of 4.2 years for the entire group.43
 OTHER THYMIC NEOPLASMS
OTHER THYMIC NEOPLASMS
Clinical assessment and management of a variety of other thymic neoplasms are considered below.
Thymic Hyperplasia
Thymic hyperplasia is a term used to describe either a histologically normal thymus that is enlarged for the patient’s age, or a thymus that histologically shows cellular hyperplasia, which is also generally associated with gross enlargement of the gland. This condition can present as a spectrum of disease ranging from respiratory compromise due to impingement on the airway (almost always in the pediatric population) to the more common situation of an incidental finding on an unrelated imaging study. Many of the thymus glands removed from patients with myasthenia gravis demonstrate “true thymic hyperplasia” (cellular hyperplasia), and this is thought to contribute to the pathogenesis of myasthenia gravis. In addition to myasthenia gravis, thymic hyperplasia can be seen following severe or chronic illness, the so-called “thymic rebound.” Steroid use will also generally lead to substantial thymic hyperplasia. Practically speaking, when a diffusely enlarged thymus without a discrete mass is discovered in an otherwise asymptomatic patient, this can be followed expectantly, as these glands have a negligible incidence of harboring significant thymic disease.44
Thymolipoma
Thymolipomas are distinguished from simple mediastinal lipomas by their location within the thymic capsule. Histologically, these neoplasms composed of mature adipocytes as well as normal thymic components.45 Thymolipomas, like thymomas, can be associated with thymic paraneoplastic syndromes such as red cell aplasia, hypogammaglobulinemia, and aplastic anemia. These lesions can grow to significant size and result in compressive and obstructive symptoms. The treatment of thyolipomas is surgical excision.
Thymic Carcinoid
Thymic carcinoids are very rare neoplasms of the thymus that are classified as neuroendocrine carcinomas. They have a strong association with endocrinopathies such as multiple endocrine neoplasia type I. Unlike other carcinoids, they rarely present with carcinoid syndrome, but many do secrete ACTH resulting in Cushing syndrome. Thymic carcinoids tend to be very aggressive tumors. Upon presentation, the majority of thymic carcinoids are locally invasive and approximately half of all patients will have metastatic disease.46 Surgery is the therapy of choice for resectable neuroendocrine carcinomas of the thymus and should include aggressive local resection. The prognosis is poor despite aggressive therapy, and most patients present with local recurrence or metastases within 5 years of surgery and will die within 10 years.47
 GERM CELL TUMORS
GERM CELL TUMORS
GCTs comprise a group of neoplasms that arise from gonadal tissue. The anterior mediastinum is the most common location for occurrence of extragonadal GCTs. GCTs account for 15% of all anterior mediastinal masses in adults (85% of which are benign mature GCTs) and 25% of anterior mediastinal masses in children (essentially all of which are benign).48 GCTs typically occur in young adults in the second to fourth decades of life with no gender predilection.
While previously thought to be metastatic from a gonadal primary, it is now accepted that these neoplasms are primary mediastinal tumors. The precise origin of the germ cells that form these tumors in sites heterotopic to the gonads is still not completely clear, but this may result from aberrant migration of germ cells during embryonic development or as a part of normal embryogenesis. Because primordial germ cells are undifferentiated cells from which a variety of different tumors can arise, there have been numerous overlapping and sometimes confusing classification and grading systems proposed for these tumors. Even more confusing for classifying GCTs is that it is common to see two or more GCT histologies arising from within a single mass. Based on the need for standard classification, investigators from the Armed Forces Institute of Pathology developed a reproducible histologic classification system (Table 82-4) and clinical staging system (Table 82-5) based on review of more than 300 cases.49



