9 Basic Physiology and Hemodynamics of Cardiac Pacing
 Physiology of Electrical and Mechanical Activation
Physiology of Electrical and Mechanical Activation
Electrical Activation During Sinus Rhythm
The cardiac action potential originates from the sinus node, located high in the right atrium (Fig. 9-1). Its cells depolarize spontaneously and initiate the spontaneous depolarization of action potentials at a regular rate from the sinus node. This rate depends on various conditions, such as atrial stretch and sympathetic activation, but is usually between 60 and 100 beats per minute (bpm) at rest. Myocytes are electrically coupled to each other through gap junctions. These structures consist of connexin molecules and allow direct intercellular communication.1 Gap junctions do not have a preferential direction of conduction, but because the action potential starts in the sinus node, it spreads from there through the atria. Evidence suggests specialized conduction pathways in the atrium, but their (patho) physiologic relevance is still disputed.
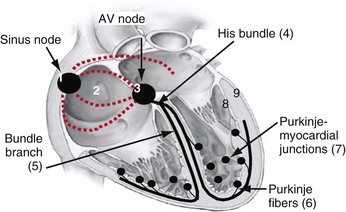
In the human heart, spreading of the action potential through the atria takes approximately 100 msec, after which the impulse reaches the AV node (see Fig. 9-1). In the normal heart the AV node is the only electrical connection between the atria and ventricles, because a fibrous ring (anulus fibrosus) is present between the remaining parts of the atria and ventricles. The AV nodal tissue conducts the electrical impulses very slowly; indeed, it takes approximately 80 msec for these impulses to travel from the atrial side to the ventricular side of the AV node. This delay between atrial activation and ventricular activation has functional importance because it allows optimal ventricular filling. Its slow conduction renders the AV node sensitive to impaired conduction and even complete conduction block, an important indication for ventricular pacing. As with other locations in the heart, conduction in the AV node has no preferential direction. Consequently, impulses can also be conducted retrogradely through the AV node, a condition that can occur when the ventricles are electrically stimulated.
From the AV node, the electrical impulse reaches the His bundle, the first part of the specialized conduction system of the ventricles called the Purkinje system. Within this system the electrical impulse is conducted approximately four times faster (3-4 m/sec) than in the working myocardium (0.3-1 m/sec). This difference results because Purkinje cells are longer and have a higher content of gap junctions.1–5
It seems that the bifascicular vision of structure of the LBB is oversimplified.6 The general picture that now emerges is that the left ventricular Purkinje network is composed of three main, widely interconnected parts, depending on the anterior subdivision, the posterior subdivision, and a centroseptal subdivision of the left main bundle. This third medial or centroseptal division supplies the midseptal area of the left ventricle and arises from the LBB, from its anterior or posterior subdivision, or both. The fascicles continue in a network of subendocardially located Purkinje fibers. In the left ventricle, Purkinje fibers form a network in the lower third of the septum and free wall, which also covers the papillary muscles.4,7 In humans the bundles are present only underneath the endocardium, whereas other species (e.g., ox, sheep, goat) have networks of Purkinje fibers across the entire ventricular wall.8,9
It is important to note that the His bundle as well as the right and left bundle branches and their major tributaries are electrically isolated from the adjacent myocardium. The only sites where the Purkinje system and the normal working myocardium are electrically coupled are the Purkinje-myocardial junctions. The exits of the Purkinje system are located in the subendocardium of the anterolateral wall of the right ventricular (RV) and the inferolateral left ventricular (LV) wall4,10 (see Fig. 9-1). This area of exit corresponds with the area of the ventricular muscle that is activated earliest.2–5,11,12 The distribution of the Purkinje-myocardial junctions is spatially inhomogeneous, and the junctions themselves have varying degrees of electrical coupling.13
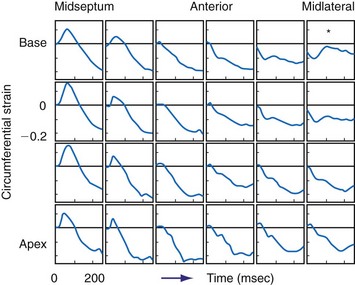
Endocardial activation of the right ventricle starts near the insertion of the anterior papillary muscle 10 msec after onset of LV activation11 (Fig. 9-2). After activation of the more apical regions, the activation of the ventricular working myocardium occurs predominantly from apex to base, both in the septum and in the LV and RV free wall. Further depolarization occurs centrifugally from endocardium to epicardium as well as tangentially.11,12 The earliest epicardial breakthrough occurs in the pretrabecular area in the right ventricle, from which there is an overall radial spread toward the apex and base. The last part of the right ventricle that becomes activated is the AV sulcus and pulmonary conus. Overall, the posterobasal area of the left ventricle (or an area more lateral) is the last part of the heart to be depolarized (see Fig. 9-2).
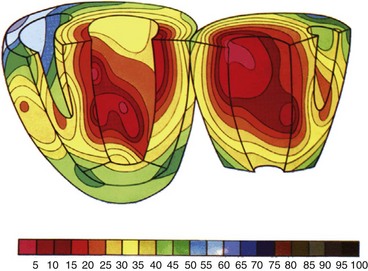
The time between arrival of the impulse in the His bundle and the first ventricular muscle activation is approximately 20 msec,4 whereas total ventricular activation lasts 60 to 80 msec, corresponding to a QRS duration of 70 to 80 msec.11 These numbers illustrate the important role of the Purkinje fiber system in the synchronization of myocardial activity. This role results from the system’s unique propagation properties and its geometrically widespread distribution. During normal orthodromic excitation, fast propagation over long fibers, together with wide distribution of Purkinje-myocardial junctions, induces a high degree of coordination between distant regions of the myocardium.
Mechanical Activation During Sinus Rhythm
As in many other muscle cells, electrical activation leads to contraction in cardiac myocytes, a process referred to as excitation-contraction (E-C) coupling. In cardiac E-C coupling, the calcium ion (Ca2+) plays a central role14 (Fig. 9-3). The contraction-relaxation cycle starts when depolarization leads to entry of Ca2+ into the cell through voltage-dependent L-type Ca2+ channels. This Ca2+ entry triggers a much larger amount of Ca2+ to be released from the sarcoplasmic reticulum (Ca2+-induced Ca2+ release). These processes increase intracellular Ca2+ from approximately 10−7 M to 2 × 10−6 M. This high calcium concentration catalyzes the interaction between myosin and actin filaments, leading to contraction. After repolarization, relaxation occurs as Ca2+ dissociates from the contractile apparatus and is taken up again by the sarcoplasmic reticulum through the action of sarcoplasmic reticular Ca2+–adenosine triphosphatase (SERCA). Intracellular Ca2+ homeostasis is also maintained through action of the sodium-calcium (Na+/Ca2+) exchanger (NCX). Normally, this exchanger removes Ca2+ from the cell (forward mode). Figure 9-3 also shows that the intracellular Ca2+ concentration rises rapidly after the upstroke of the action potential, but that there is some delay between the Ca2+ increase and the development of force. This delay is the main determinant of the delay between electrical and mechanical activation.
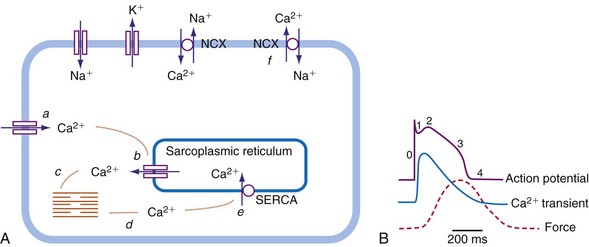
The electromechanical delay, that is, the delay between the depolarization and the onset of force development, amounts to approximately 30 msec.15,16 On a global basis, this delay can be observed as the delay between the R wave of the electrocardiogram (ECG) and the rise in LV pressure (Fig. 9-4). Electromechanical coupling in failing hearts is different from that in normal hearts. A prominent feature of heart failure is that the relation between SERCA and the NCX changes. Often, SERCA is downregulated17 and NCX upregulated in the heart failure state.18 On the one hand, the reduced SERCA activity leads to less Ca2+ loading of the sarcoplasmic reticulum, resulting in less Ca2+ release during the subsequent activation, and thus a weaker contraction (systolic dysfunction). On the other hand, during diastole, Ca2+ removal from the cytosol is slower and incomplete, leading to a slower relaxation and greater diastolic stiffness (diastolic dysfunction). The upregulation of the NCX and the elevated intracellular Na+ concentrations in failing myocardium19 may compensate for the SERCA downregulation.20,21 The high Na+ concentration facilitates the NCX to work in the “reverse mode,” that is, to remove intracellular Na+ and exchange it for extracellular Ca2+. This additional Ca2+ enhances systolic function at least to some extent. Because the SERCA pump is much faster in pumping calcium than the NCX, insufficiency in contraction and relaxation in failing hearts is most pronounced at higher heart rates. These changes result in decreasing contractile force with increasing heart rate, a negative force-frequency relation.17 The changes in the failing myocardium lead to the altered expression of other proteins involved in E-C coupling, as well as shifts in isoforms of various contractile proteins.22
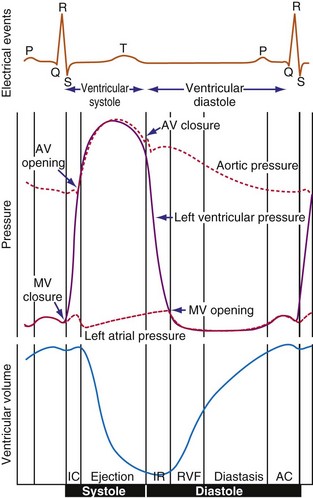
Because of the tight coupling between excitation and contraction, atrial activation is followed by atrial contraction, and ventricular activation by ventricular contraction. Consequently, atrial contraction precedes ventricular contraction, as illustrated in Figure 9-4. Because of this timing, the atrial contraction adds about 20% to the volume of the ventricles. This “atrial kick” increases the length of ventricular muscle cells and their sarcomeres. The sarcomere length is an important determinant of myocardial contractile force. Over the entire physiologic range of sarcomere lengths (1.6-2.4 µm), the longer the sarcomeres, the greater is the contractile force. This effect, known as the Frank-Starling relation (Starling curve; Starling law of the heart), is only partly related to the overlap of actin and myosin, as generally mentioned in textbooks. More important to the Frank-Starling relation is the growing affinity of troponin C for calcium at increasing sarcomere length,23 although the cause of this greater sensitivity is not completely understood.
Ventricular contraction starts after ventricular depolarization. After a short isovolumic contraction phase, the aortic valve opens, starting the ejection phase. The velocity of emptying of the ventricle is highest in the first half of the ejection phase because of a higher pressure gradient between the left ventricle and the aorta. In the second half of the ejection phase, LV pressure actually falls below aortic pressure, but the aortic valve stays open because of the inertia of the flowing blood. With increasingly negative LV-aortic pressure gradients, the aortic valve closes and the isovolumic relaxation phase starts, ending with the opening of the mitral valve. Because of the filling of the atrium during ventricular systole, atrial pressure is relatively high. This event, in combination with the rapid fall in LV pressure, causes a positive AV pressure gradient during the early filling phase. Thus, rapid acceleration of blood occurs, contributing to most of the LV filling in the early diastolic phase. This event is also reflected by the large Ei wave on mitral valve Doppler recordings in healthy hearts (Fig. 9-5).
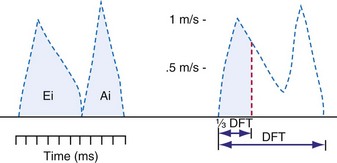
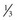 DFT [diastolic filling time]).
DFT [diastolic filling time]).Abnormal Activation Sequence During Left Bundle Branch Block and Right Ventricular Pacing
The normal, physiologic, and almost synchronous sequence of electrical activation is lost in diseases affecting the ventricular conduction system, such as block of the left or right bundle branch24 and the presence of an accessory pathway bypassing the AV node, as in the Wolff-Parkinson-White syndrome.25 Additionally, ectopic impulse generation, occurring during ventricular pacing and extrasystoles,26,27 leads to abnormal impulse conduction. Under all these circumstances, the impulse is conducted primarily through the slowly conducting working myocardium rather than rapidly through the specialized conduction system. As a consequence, under conditions of abnormal activation, the time required for activation of the entire ventricular muscle, expressed as QRS duration, is at least twice as long as that during normal sinus rhythm.
Detailed studies on the three-dimensional spread of activation during ventricular pacing in canine hearts have been conducted since the 1960s.12,26 The sequence of electrical activation in LBB block (LBBB) is similar to that during RV apex pacing. This sequence can be derived from the QRS configuration of the surface ECG27,34,35 and from endocardial activation maps in experimental LBBB and RV pacing.36 The similarity has lead to the use of AV sequential RV apex pacing as a model for “experimental LBBB.”34,37,38
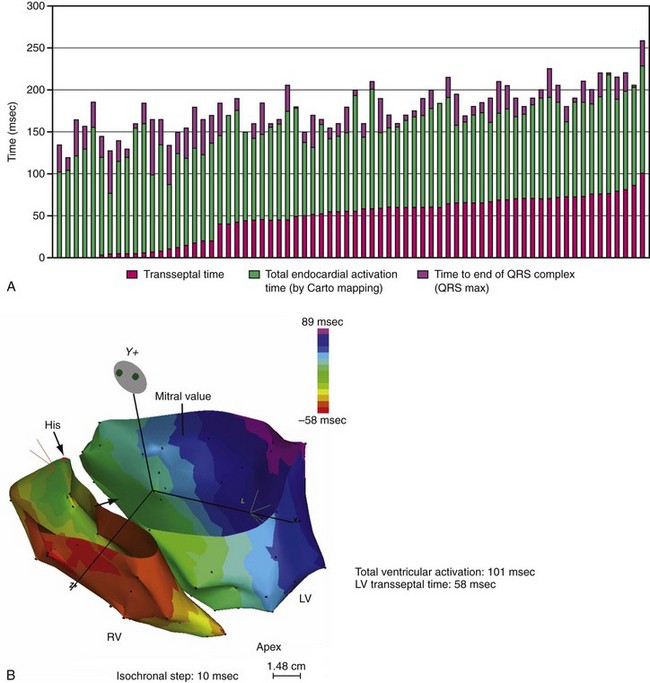
Until recently, information on impulse conduction in patients with LBBB was limited to the data reported by Vassallo et al.24,27 These investigators mapped LV endocardial activation during RV apex pacing and in LBBB at a limited number of endocardial sites, showing that activation starts at the RV endocardium in both conditions. The first noticeable activation at the LV endocardium after right-to-left conduction of the impulse occurs at a single breakthrough site, which in almost all patients is the LV breakthrough time, 50 to 70 msec after the earliest RV activation. The impulse is conducted from the septum toward the distal free wall in a gradual manner, the site of latest activation generally being the inferoposterior wall.11 More recently, this finding has been confirmed using high-resolution mapping system39–42 (Fig. 9-6). These studies also showed that activation patterns differ among patients largely because of differences in the origin of LBBB: either a proximal conduction block43,44 or a uniform but slow conduction through the LBB.45 In approximately one third of patients with heart failure and typical LBBB QRS morphology, transseptal activation time (i.e., time between earliest RV and LV septal breakthrough point) is bimodally distributed and has a large range in both groups of patients, with transseptal conduction time of 40 msec or less and longer than 40 msec.46 This indicates great heterogeneity in the RV-to-LV activation time. Furthermore, in patients with LBBB, the total LV endocardial activation time ranges from 60 to 160 msec. Etiology does not seem to have a major impact on the total endocardial activation time. Of note, the sum of transseptal and total endocardial activation times does not account for the maximum QRS duration, which is 20 to 60 msec longer, probably because of LV endocardial-to-epicardial conduction time (Fig. 9-7). These data also show that QRS duration provides a reasonable although imperfect estimate of total electrical asynchrony.
Patients with LBBB show a peculiar spread of ventricular activation.40,42 Indeed, in two thirds of patients with LBBB, a functional line of block is present, resulting in a U-shaped electrical activation wavefront. The presence and location of the line of block have been also recently observed in canine model of LBBB using the same noncontact mapping system.46a The activation front propagates first around the inferior wall and then spreads onto the lateral and basal walls. The location and length of the line of block vary greatly and are related to the site and time of LV breakthrough. In patients with significant prolongation of the QRS duration (>150 msec), the line of block is consistently located in an anterior position. In contrast, in patients with QRS duration of 120 to 150 msec, the line of block is usually shorter and is located anteriorly, laterally, or posteriorly40 (Movie 9-1).
Abnormal Contraction Patterns During Left Bundle Branch Block and Right Ventricular Pacing
Given the tight relationship between excitation and contraction in the myocardium, it is not surprising that asynchronous electrical activation also leads to asynchronous contraction. Regions where the impulse arrives first also start to contract first.47 During LBBB or ventricular pacing, local contraction patterns differ not only in the onset of contraction, but also, and more importantly, in the pattern of contraction. These contraction patterns imply that opposing regions of the ventricular wall are out of phase and that energy generated by one region is dissipated in opposite regions. In patients with LBBB or RV pacing, the early-contracting region is most often the septum. The earliest-contracting fibers can shorten rapidly by up to 10% just during the isovolumic contraction phase, because the remaining muscle fibers are still in a relaxed state (Movie 9-2). This rapid, early shortening is followed by an additional but modest systolic shortening, eventually followed by systolic stretch (from delayed mechanical contraction of other regions, in lateral wall), and premature relaxation (Fig. 9-8). In late-activated regions, in contrast, the fibers are stretched in the early systolic phase (up to 15%) from contraction of the early-activated region. Doubling of net systolic shortening and delayed relaxation occur in late-activated regions.31,48 The discoordination between early- and late-activated regions leads to lower output and reduced efficiency of the heart as a pump.
The regional differences in contraction pattern most likely results from the local differences in myocardial fiber length during the early systolic phase. This is supported by studies using two isolated papillary muscles in series in which asynchronous stimulation caused a downward shift in the force-velocity relation in the earlier-activated muscle and an upward shift in the later-activated one.49 Furthermore, during ventricular pacing, regional systolic fiber shortening increases with greater isovolumic stretch.50 Similarly, a close correlation exists between the time of local electrical activation and the extent of systolic fiber shortening.51 Therefore, the regional differences in contraction pattern during ventricular pacing are most likely caused by regional differences in effective preload and local differences in the contraction force triggered by the Frank-Starling relation.
On M-mode echocardiography, abnormal contraction patterns during RV pacing and LBBB appear as paradoxical septal motion.52 However, this motion is not actually paradoxical, because it is really the net result of different forces. The motion is caused by the asynchrony between RV and LV, which produces dynamic alterations in transseptal pressure differences,52,53 and by presystolic shortening of septal muscle fibers.54 Abnormal septal motion results in a diminished contribution of the interventricular septum to LV ejection. This has been measured in experimental LBBB using MRI tagging54 and in patients with LBBB QRS morphology using speckle tracking.55,56 The septal regions, even in absence of detectable scar, showed the lowest amount of systolic strain within LV; in contrast, the lateral wall shows the highest regional strain.55 This finding demonstrates that LBBB determines a unique and unequal LV strain distribution that may be corrected by CRT.55–57
Effect of Left Bundle Branch Block and Right Ventricular Pacing on Local Energetic Efficiency
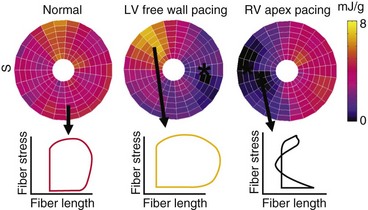
The local differences in wall motion and deformation mentioned previously reflect regional differences in myocardial work.48,58 This relationship was demonstrated by construction of local fiber stress–fiber strain diagrams and calculation of local external and total mechanical work. In regions close to the pacing site (or septum in patients with LBBB), shortening occurs at low pressure, whereas these areas are transiently being stretched at higher ventricular pressures. As a consequence, the stress-strain loops have a figure-8-like shape with a low net area, indicating the absence of external work. In regions remote from the pacing site (or lateral wall in patients with LBBB), the loops are wide, and external work can be up to twice that during synchronous ventricular activation (Fig. 9-9). Total myocardial work (sum of external work and potential energy) in LBBB and with RV pacing is reduced by 50% in early-activated regions and is increased by 50% in late-activated regions in comparison with atrial pacing.48,54,59
Several studies reported that ventricular pacing and LBBB are associated with regional differences in myocardial blood flow,31,35,38,54,58–60 glucose uptake,38,61 and oxygen consumption.58,62 During RV apex pacing and LBBB, low values are found in the septum, the early-activated part of the left ventricle. Compared with sinus rhythm, myocardial blood flow and oxygen consumption are 30% lower in early-activated regions and 30% higher in late-activated regions.31,58,59 Controversy surrounds the cause of the reduced septal blood flow and glucose uptake in LBBB and RV apex pacing. Various observations, such as close correlations among local myocardial oxygen consumption, work, and blood flow, suggest that the lower blood flow is a physiologic adaptation to the lower demand. However, at higher heart rates, the asynchronous contraction pattern may also impede myocardial perfusion.38,63 In patients with RV pacemakers, false-positive findings of perfusion defects may result from this redistribution.64
The nonuniform contraction patterns translate into a lower efficiency of conversion of metabolic energy into mechanical pump function. This was observed in isolated, isovolumically beating hearts,65 as well as in anesthetized open-chest,66,67 closed-chest,59 and conscious dogs.68 Mills et al.59 also showed that efficiency remained low after 4 months of RV pacing. In all these preparations, RV apex pacing reduced mechanical output, whereas myocardial oxygen consumption was unchanged or even increased compared with atrial pacing; thus efficiency decreased by 20% to 30% in these studies. The opposite change was found when ventricular activation in patients with preexisting dyssynchrony caused by LBBB-like activation was improved by biventricular (BiV) or LV pacing alone. In these patients, BiV pacing improved the maximal rate of rise of LV pressure (LV dP/dtmax) without increasing myocardial oxygen consumption, indicating improved efficiency.69 Efficient conversion of metabolic energy to pumping energy is of particular interest in patients with compromised coronary circulation, because higher oxygen consumption over longer periods (as in β-agonist therapy) leads to higher mortality.
Effect of Asynchronous Activation on Systolic and Diastolic Pump Function
Both RV pacing and LBBB reduce systolic and diastolic function. These effects are independent of changes in preload and afterload. This conclusion is based on results from studies using preparations in which preload and afterload were controlled,65 as well as those in which preload- and afterload-independent indices of ventricular function were determined68,70–72 (Figs. 9-10 and 9-11). The negative mechanical effect of dyssynchronous activation has been observed under various loading conditions73 and during exercise.74 Ventricular pacing also causes a deterioration in pump function in patients with coronary artery disease,75 as well as those with already-impaired LV function76 (Fig. 9-12). Impaired regional and global cardiac pump function has been observed in patients and animals with LBBB, even if LBBB was not accompanied by other cardiovascular diseases.53,54 Therefore, it appears that under all circumstances, dyssynchrony is an important, independent determinant of cardiac pump function.
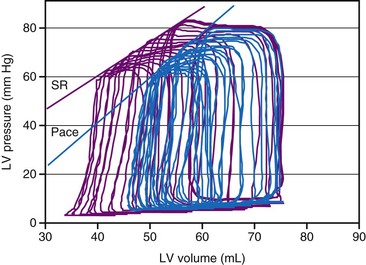
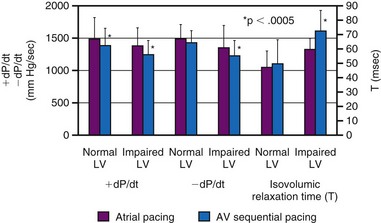
Although LV dP/dtmax is a preload-dependent parameter, it is also sensitive to changes in ventricular activation sequence, as shown both in animal models and in humans34,76–78 (see Fig. 9-12). Thus, LV dP/dtmax appears to be an appropriate marker of dyssynchrony-induced changes in LV global contractile function and its correction by any pacing technique. Additionally, the rate of ventricular relaxation is slower during ventricular pacing, with more pronounced detrimental effect on relaxation, in patients with impaired ventricular function76 (see Fig. 9-12). Isovolumic relaxation parameters, such as LV dP/dtmin and Tau, are strongly influenced by pacing.78–80 Parameters of auxotonic relaxation (rate of segment lengthening or LV volume increase) are also lower during ventricular pacing than during sinus rhythm, but the difference is less pronounced than for the isovolumic relaxation parameters.73,78
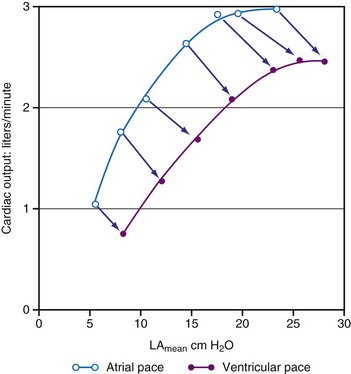
As a consequence of the slower contraction and relaxation, isovolumic contraction and relaxation phases last longer, thus leaving less time for ventricular filling and ejection.53,65,81 Therefore, it is not surprising that cardiac output and systolic arterial and LV pressures are also affected by a dyssynchronous activation. In general, stroke volume is affected more than systolic LV pressure,72,77 presumably because baroreflex regulatory mechanisms partly compensate the decrease in blood pressure. This idea is supported by the finding of higher catecholamine levels82 and greater systemic vascular resistance74 during ventricular pacing. With regard to the changes in stroke volume, it is important to note that ventricular pacing, especially RV apex pacing, can induce mitral regurgitation, as has been demonstrated in animals83,84 and patients.85,86 In addition to reduced stroke volume at unchanged preload, ejection fraction is usually found to be depressed during ventricular pacing74,87 as well as in LBBB.88 Similarly, ventricular pacing can increase pulmonary wedge pressure.74 The negative inotropic effect of ventricular pacing under various loading conditions is clearly illustrated by a rightward shift of the LV function curve, that is, the relationship between cardiac output and mean atrial pressure71 (see Fig. 9-10). Later studies showed a rightward shift of the end-systolic pressure-volume (P-V) relation (see Fig. 9-11), thus suggesting that for each end-systolic pressure, the LV must operate at a larger LV volume.70,72,89
Cause of Reduced Pump Function
An obvious cause of reduced pump function during abnormal electrical activation is the asynchronous contraction of the different parts of the ventricular muscle. Using a mathematical simulation, Suga et al.90 divided the LV wall into two elements with similar contractile properties. Contractility of the whole left ventricle decreased considerably when the asynchrony exceeded 100 msec. This concept is supported by experimental observations in dogs with LBBB. In studies using combinations of pacing sites, minimal intra-LV asynchrony, assessed by endocardial electrical activation mapping, consistently led to the highest LV dP/dtmax.36 In addition to intraventricular asynchrony, interventricular asynchrony may affect ventricular pump function. An extensive overview of studies on RV and LV pacing showed that RV pacing consistently reduces LV function more than LV pacing.91 Even at similar levels of intraventricular asynchrony, LV dP/dtmax is lower during RV pacing than during LV pacing.36 This suggests important roles for interventricular asynchrony and associated interventricular mechanical interaction. This ventricular interaction is also expressed by abnormal septal motion in asynchronous ventricles.52
Also, the pathway of activation may be a determinant of ventricular function. The sequence of activation from apex to base appears to be important. As already mentioned, the ventricular myocardium is activated from apex to base during normal sinus rhythm. Among all LV pacing sites, the LV apex appears to keep LV pump function closest to that seen during atrial pacing.26,59,92–94 The observation that the addition of pacing sites during LV apex pacing does not improve and sometimes even reduces LV function emphasizes the importance of a ventricular activation directed from apex to base.94
An additional cause of the reduction in pump function during asynchronous activation is mitral valve insufficiency.83,85 This diminishes LV pump function directly by reducing the volume ejected into the aorta and indirectly by reducing LV cavity volume. The immediate cause of mitral valve regurgitation during RV apex pacing is papillary muscle desynchronization.95 In addition, papillary muscle tethering forces may be increased,96 and the transmitral pressure gradient may be decreased.97
Electrical and Structural Remodeling
Asynchronous electrical activation not only has acute mechanical consequences, but also influences cardiac structure and function over time. A particular adaptation of the heart to ventricular pacing occurs when the abnormal activation created by pacing is stopped but repolarization remains abnormal, a phenomenon called cardiac memory.98 Several investigators found evidence that, as in the nervous system, short-term cardiac memory (<1 hour) involves changes in ion channels and phosphorylation of target proteins,99 and that long-term cardiac memory (≈3 weeks of pacing) involves altered gene programming and protein expression.98,100 Interestingly, in humans, cardiac memory appears to reach a steady state within 1 week,101 which appears to be quicker than in dogs.98 Costard-Jäckle and Franz102 have demonstrated opposite changes in repolarization in regions remote from and close to a pacing site starting within 1 hour of ventricular pacing.
The repolarization abnormalities related to cardiac memory also have a mechanical counterpart. During sinus rhythm immediately after a period of ventricular pacing, relaxation is disturbed.103 Moreover, systolic function deteriorates between 2 hours and 1 week after ventricular pacing has been stopped (Fig. 9-13). After ventricular pacing is stopped for 1 week, it takes 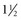 days for ejection fraction to return to its prepacing value.104 It seems likely that changes in the function of an ion channel, such as the L-type calcium channel,99 underlie the reduction in ejection fraction during the first week of ventricular pacing. Therefore, electrical and contractile remodeling appears to occur soon after the onset of asynchronous activation.
days for ejection fraction to return to its prepacing value.104 It seems likely that changes in the function of an ion channel, such as the L-type calcium channel,99 underlie the reduction in ejection fraction during the first week of ventricular pacing. Therefore, electrical and contractile remodeling appears to occur soon after the onset of asynchronous activation.
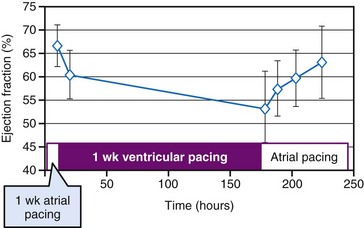
In addition to the long-term effect on cardiac memory, longer-lasting (>1 month) ventricular pacing and LBBB lead to major structural changes, such as ventricular dilatation and asymmetrical hypertrophy.54,70,105 The ventricular dilatation appears related to the LV operating at a larger volume. Global hypertrophy may be induced by this global dilatation, as well as by the greater sympathetic stimulation82 (Fig. 9-14). The asymmetry of hypertrophy appears to be more prominent during LV pacing than during RV pacing and LBBB.54,70,106 However, regardless of pacing site and underlying conduction disturbance, the asymmetry of hypertrophy is characterized by a more pronounced growth of the late-activated myocardium, that is, the same region that shows enhanced contractile performance because of early systolic prestretch. It has been observed in juvenile canine hearts that ventricular pacing leads to fiber disarray,107 which may also be the result of the regionally different and abnormal distribution of mechanical work.31,48,58,59,108
In ventricles undergoing long-term pacing,70 myocyte diameter is increased in late-activated regions, but the number of capillaries per myocyte is unchanged (van Oosterhout and Prinzen, unpublished data). Therefore, the diffusion distance for oxygen increases, potentially leading to compromised oxygenation, which is known to render hypertrophic myocardium susceptible to ischemia. Resynchronization has been shown to result in improved coronary reserve, suggesting that the structural remodeling at the microvascular level may also be reversible.109 An alternative explanation is that CRT could be simply decreasing the oxygen demand in the late-activated regions.
In conditions such as infarction and hypertension, ventricular remodeling appears, at least initially, to be meant to compensate for the loss of function and the increased load, respectively. However, dilatation and hypertrophy do not reduce the asynchronous activation induced by pacing or LBBB; if any, they increase it.54,70 The reasons are the longer path length of impulse conduction (dilatation) and the larger muscle mass to be activated (hypertrophy). Moreover, chronic asynchronous activation also reduces expression of gap-junction channels110 and induces lateralization of these channels.111 Akar et al.112 have related the reduced expression in nonfailing and failing asynchronous hearts to slower impulse conduction, especially in the late-activated regions. Moreover, they showed that action potential duration increases in the late-activated myocardium. These molecular changes may render the myocardium not only more asynchronous, but also more susceptible to arrhythmias.
Even more complicated molecular changes occur in animal models with combined asynchronous activation and heart failure, such as that caused by rapid ventricular pacing. Under these conditions, beta-adrenergic receptors are downregulated,113 as are many ion channels, such as most potassium and calcium channels, whereas other channels display a regional difference in expression, such as the L-type calcium channel.114 Moreover, in both patients with LBBB115 and animal models of dyssynchrony,113 several proteins related to or facilitating the apoptotic process show unique regional distribution, further confirming the hypothesis that abnormal electrical activitation sequence may contribute to remodeling in heart failure patients.
All these processes may lead to a vicious circle in which dilatation and hypertrophy further reduce LV pump function, either directly or by increasing asynchrony of activation (see Fig. 9-14). As is the case after myocardial infarction, initial compensatory hypertrophy may result in heart failure many years later. Clinical evidence for such an important role of asynchronous activation in the development of heart failure is discussed later.
 Physiologic Effects of Heart Rate and Atrioventricular Synchrony
Physiologic Effects of Heart Rate and Atrioventricular Synchrony
Correction of Bradycardia by Pacing
Cardiac pacing is the only effective treatment for symptomatic bradycardia. Permanent pacemakers have been implanted since the early 1960s to prevent death or syncope caused by ventricular asystole. Although asystole is clearly to be prevented, bradycardia does not always immediately lead to pump failure. This fact can be derived from studies on young patients with congenital AV block. As a compensation for the bradycardia, these patients have enlarged hearts and an increased ejection fraction.116,117 In a 10-year follow-up study, 9 of 149 patients demonstrated dilated cardiomyopathy.117
At the onset of acquired complete heart block, cardiac output is smaller than with normal heart rate.118 As a result, compensatory increases in sympathetic tone and end-diastolic ventricular volume occur, leading to higher atrial rates, enhanced ventricular contractility, and augmented stroke volume. Over the short term, the canine heart adapts to bradycardia by increasing diastolic volume and neurohumoral activation, which results in BiV hypertrophy118–120 (Table 9-1). Although these adaptations initially manage to return cardiac output to the level before the onset of AV block, this compensatory effect usually fades after 2 months of heart block. After 4 months, contractility is back to baseline, but LV cavity volume and corrected QT interval (QTc) continue to increase118 (Fig. 9-15); the latter increase is associated with an enhanced susceptibility to arrhythmias.119 If complete AV block persists for more than 4 months, congestive heart failure (CHF) ensues.120 Development of CHF may originate from the ventricular dilatation and hypertrophy initiated early after the start of heart block.
TABLE 9-1 Hemodynamic Effects of Ventricular Pacing in Complete Heart Block (CHB)
| Hemodynamic Parameter | CHB with Slow Ventricular Response | Ventricular Pacing in CHB |
|---|---|---|
| Ventricular rate | ↓ | ↑ |
| Cardiac index | ↓ | ↑ |
| Stroke volume | ↑ | ↓ |
| Sympathetic tone | ↑ | ↓ |
| Ventricular contractility | ↑ | ↓ |
| Systemic vascular resistance | ↑ | ↓ |
| Atrial rate | ↑ | ↓ |
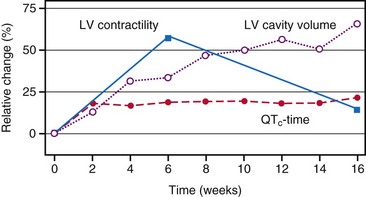
The importance of the duration of overload is confirmed by the observation that the frequency of CHF symptoms in patients with complete heart block correlates with the duration of heart block. CHF has been described during chronic complete heart block even in patients with normal ventricular function. Brockman and Stoney121 reported that in two thirds of their patients with complete heart block and CHF, symptoms were relieved by ventricular pacing alone, and no additional medical therapy was required. Breur et al.122 showed that, in patients with congenital heart block, implantation of a pacemaker resulted in less LV dilatation than in the untreated control group, in which the LV cavity gradually dilated. Similarly, in dogs with AV block, LV dilatation gradually progresses over time (see Fig. 9-15), whereas ventricular pacing readily reverses the LV dilatation and hypertrophy caused by AV block.93 However, longer-lasting ventricular pacing in hearts with AV block may lead to LV dilatation and hypertrophy secondary to the effect of asynchronous activation. Two studies on young adults who received pacing for approximately 10 years showed a high preponderance of CHF and LV dilatation.123,124 These data strongly suggest that in the patient with AV block, the short-term and long-term effects of RV pacing are different. Therefore, the decision whether or not to start pacemaker treatment using RV pacing or the choice of the pacing site should be made carefully (see later).
Early studies suggested that the maximal increase in cardiac output during ventricular pacing at rest occurs at rates between 70 and 90 bpm125–127 (Fig. 9-16). Further rises in rates result in either no additional increase or a decrease in cardiac output accompanied by greater peripheral vascular resistance. In contrast, there is growing evidence that lower heart rates may be particularly beneficial. Indeed, in animal models it appears that bradycardia promotes angiogenesis.128,129 Moreover, Lei et al.130 demonstrated that reducing normal heart rates by 20% to 30% through the continuous infusion of alinidine in a post–myocardial infarction (post-MI) rat model increased basic fibroblast growth factor, its receptor, and expression of related proteins. Bradycardia in this model also increased capillary length density in the border zone of the infarct by 40% and in the remote zone by 14%. Bradycardia also increased arteriolar density in the septum by 62%. These changes translated to a 23% greater coronary reserve, a smaller increase in post-MI left LV volume, and a greater preservation of ejection fraction in the bradycardic animals.
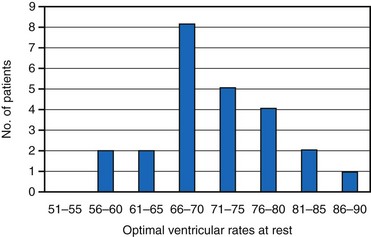
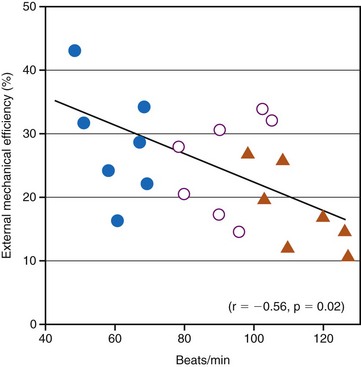
Although the relationship between optimal resting ventricular pacing rate and ventricular dysfunction has not been systematically evaluated, it is likely that different resting ventricular rates are required in patients with systolic and diastolic dysfunction. For example, Ishibashi et al.131 reported that in elderly hypertrophic patients, even 25% increases in heart rate, as induced by atrial pacing, significantly reduced cardiac mechanical efficiency. The investigators went as far as to suggest that coronary perfusion itself in elderly hypertensive hypertrophic patients is negatively affected by an increase in heart rate (Fig. 9-17). They recommended that in the treatment of elderly hypertensive patients, the control of the heart rate in addition to the control of blood pressure might be helpful in minimizing the occurrence of myocardial ischemia and might slow down the subsequent progression to heart failure. Since then, their recommended treatment has become common practice with the use of β-blocker therapy to prevent MI and heart failure.
The importance of increasing heart rate to augment cardiac output with exercise has been clearly documented in several studies.132–134 Because the improvement in exercise performance is caused predominantly by an increase in heart rate, it is expected that favorable hemodynamic results will be obtained by rate-adaptive systems. Consequently, the modern approach to the management of complete symptomatic heart block involves the use of pacemaker units that provide not only basic ventricular pacing support, but also adaptation of the pacing rate to physiologic needs.
A major concern, especially in compromised hearts, is that heart rate should not increase too much, because shortened diastolic filling time, reduced LV compliance at higher rates of ventricular pacing, and increased systemic vascular resistance may limit cardiac output.116,126 Rowe et al.135 found other reasons not to increase pacing rate too greatly. They examined cardiac output and coronary blood flow at low (47 bpm), intermediate (77 bpm), and high (117 bpm) ventricular pacing rates in patients with complete heart block. Cardiac output rose with an increase from low to intermediate pacing but diminished at the high rate. However, coronary blood flow and cardiac oxygen consumption rose progressively with increasing heart rates, thus indicating lower pumping efficiency at the high rate.
Optimal Atrioventricular Interval
Importance of Atrioventricular Synchronization
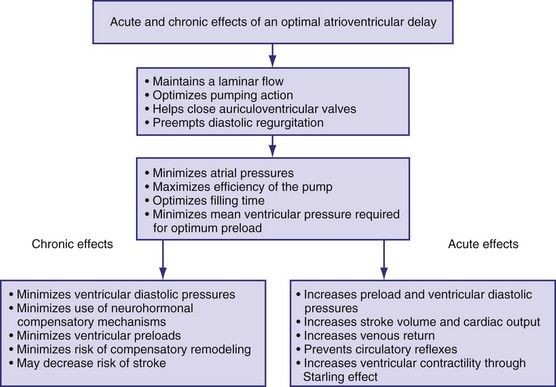
Basically, the role of the atrial contraction is to help maintain a laminar flow of blood from the venous system to the ventricles; this role is tightly integrated with the movement of the ventricles to create a smooth motion of blood across the system. To perform its role, the atrium changes its function from that of a conduit during early filling to that of a booster pump during atrial systole, to that of a reservoir during ventricular systole. These changes are closely related to longitudinal displacement of the anulus of the mitral and tricuspid valves along the long axis of the heart. Indeed, the anulus is displaced toward the apex of the ventricles during systole and toward the atria during diastole, thus significantly contributing to the smooth movement of blood from the atria to the ventricles. This displacement keeps the overall volume of the heart almost constant during systole and diastole.136
In the early years of pacing, the electrical stimulus was selectively applied to the ventricle (ventricular single-chamber pacing, or V pacing). With V pacing, the contraction of atria and ventricles is uncoupled, leading to an atrial contribution to LV filling that varies from beat to beat. This also results in large beat-to-beat variations in stroke volume, systolic pressure, and other hemodynamic variables.137 The introduction of sequential AV pacing resulted in more regular heart beats and improved hemodynamics in both animals78,138,139 and patients.140 With advances in pacemaker technology, appreciation of the importance of maintaining AV synchrony has improved, and programming of optimal AV intervals in patients with dual-chamber pacemakers is of great importance.141
Figure 9-18 summarizes the main consequences of an optimal AV timing. Because filling time is limited, especially at high heart rates, the atrial contribution to stroke volume is more prominent at high than at low heart rates.142
Patients with Normal Left Ventricular Function
The AV interval that maximizes resting cardiac output during dual-chamber pacing varies widely among patients, usually reported as 125 to 200 msec. In patients with pacemakers, cardiac output increased by 4% to 20% when the AV interval was lengthened from 0 to between 100 and 130 msec.140,143–145
Optimal AV interval can be determined in most patients during dual-chamber pacing by means of various invasive and noninvasive measurements. Most often, optimal AV interval is assessed by LV outflow recording with Doppler echocardiography (Fig. 9-19). Doppler-derived cardiac outputs at varying AV intervals can be used to fine-tune the AV interval. In patients with preserved ventricular function, this method is highly reliable, is sensitive to small output changes, and has a low interobserver variability. The optimal AV interval determined with Doppler echocardiography correlates well with the optimal AV interval determined with radionuclide ventriculography. Factors that may influence the optimal AV interval in different patients are summarized in Table 9-2. In addition to interpatient variability, other factors may influence determination of optimal AV interval in the same patient, such as heart rate, paced or sensed atrial event, posture, and drugs.146–150 Moreover, in a specific patient’s lifetime, there may be situations in which the optimal AV interval is different from the AV interval considered to be physiologic. For example, AV sequential pacing for complete heart block complicating an acute MI or after cardiac surgery yields optimal hemodynamics at AV intervals in the range of 80 to 120 msec. This may be related to a surge of intrinsic catecholamine levels or to administered inotropic drugs and reduced LV compliance in these acutely stressful situations.
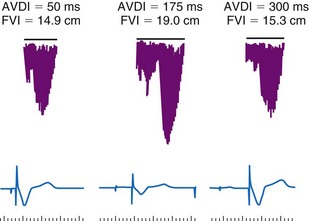
TABLE 9-2 Possible Factors Influencing Optimal Atrioventricular Intervals
| Type | Variables |
|---|---|
| Interpatient | |
| Intrapatient |
The programmed AV interval regulates the timing of right atrial (RA) and RV conduction, which in a normal heart without significant atrial electrical disease usually coincides with that of left atrial (LA) and LV conduction. From a hemodynamic point of view, LA and LV conduction is most important, followed by the left-sided AV interval. The physiologic left-sided AV interval depends on several parameters, including the programmed AV interval, latency in atrial capture and sensing, interatrial conduction time, latency in ventricular capture, and interventricular conduction time.151 Because of the complex relationship among these intervals (see Fig. 9-19), significantly different right-sided and left-sided AV intervals in the same patient and in different patients can be recorded. Among these times, the one that most influences the optimal left-sided AV interval is the interatrial conduction time, measured as the time from the atrial pacing spike to the atrial depolarization on the esophageal electrogram or to the A wave on Doppler echocardiography. Patients with near-normal interatrial conduction time (≤90 msec) derive the greatest hemodynamic benefit from programmable AV intervals of about 150 msec; in contrast, in those patients presenting with prolonged interatrial conduction time, the AV interval should be set at 200 msec or longer to maximize cardiac output. Of particular note, programming a short AV interval in patients with prolonged interatrial conduction delay may result in depolarization of the LA after LV mechanical activation. This may produce hemodynamic values equivalent to or worse than those seen in patients with VVI pacing alone. Pacemaker syndrome may also occur under these circumstances.
Latency in atrial capture and sensing may lead to different optimal AV intervals for sensed P waves and paced P waves (Fig. 9-20). The magnitude of atrial capture and sensing latencies varies among patients and may be affected by the lead and pacemaker circuitry characteristics, electrode position, tissue interface, amplitude and rate of stimulation, P-wave morphology, myocardial disease, electrolytes and other metabolic factors, and drugs. The optimal AV interval for a sensed P wave is about 30 to 40 msec shorter than the optimal AV interval for a paced P wave followed by a paced QRS at a similar heart rate (Fig. 9-21). However, differences between optimal AV intervals for a paced P wave and a sensed P wave may be as great as 100 msec in some individuals. New dual-chamber pacemakers incorporate programmable features designed to adjust the AV interval automatically in response to paced versus sensed P waves and in response to the atrial rate. It is important to recognize that the physiologic AV interval is never equivalent to the programmed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


