Arterial Disease—Atherosclerosis
Heba Wassif MD, MPH
Frederick GP Welt MD
There is increasing understanding of the molecular and cellular pathophysiology of the vascular responses to injury that lead to coronary atherosclerosis and the clinical sequelae of the disease, whether exertional angina or acute myocardial infarction (MI). A common thread that links these events is an inflammatory response to injury, and it is increasingly appreciated that the inflammatory process not only initiates these lesions but often dictates their clinical presentation. A fundamental knowledge of these processes is necessary to understand the natural history of the disease processes that affect the patients who present to the catheterization laboratory and, in addition, to understand the consequences of the therapies employed during coronary intervention. This chapter is therefore designed to describe the known pathophysiology of atherosclerosis, and the conversion of stable atherosclerotic plaques to ones that cause acute coronary syndromes (ACS).
ATHEROSCLEROSIS: A RESPONSE TO INJURY
Atherosclerosis is a chronic inflammatory disease initiated and sustained by injury to the vascular wall (1). Largely through extensive epidemiologic studies, several injurious processes have been identified (Table 1-1). These include metabolic conditions such as sustained exposure to low-density lipoprotein (LDL), hyperglycemia associated with diabetes, and hyperhomocysteinemia. However, other factors, including physical (hypertensive changes in shear stress), environmental (tobacco smoke), and possibly infectious (Chlamydia pneumoniae or viral entities) processes, have also been implicated. The common thread of injury to the vessel wall is an inflammatory response that involves a complex and still incompletely understood sequence of interactions between endothelial and smooth muscle cells (SMCs), leukocytes, and platelets. These cells and their secreted growth factors and cytokines combine with lipids and components of the vessel wall to eventually form the mature atherosclerotic plaque. The central role of inflammation in the pathogenesis of atherosclerosis is evidenced by numerous epidemiologic studies that demonstrate a correlation between circulating markers of inflammation (e.g., fibrinogen, C-reactive protein [CRP], serum amyloid protein, and myeloperoxidase) and subsequent risk of coronary events (2, 3).
TABLE 1-1 Causes of Vascular Injury | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
ATHEROSCLEROSIS: PATHOGENESIS
There are several key biologic events involved in atherogenesis: extracellular lipid accumulation, leukocyte recruitment, foam cell formation, neointimal growth as a result of SMC migration and proliferation, extracellular matrix deposition, and vessel remodeling (Fig. 1-1).
Extra- and Intracellular Lipid Accumulation
The key event in the creation of the incipient atherosclerotic lesion is the accumulation of lipoproteins within the intima. These lipoproteins may subsequently be modified by processes such as oxidation and glycation under the conditions of aging or hyperglycemia. The modification of these lipoproteins helps to elicit a cascade of molecular and cellular events, including stimulation of growth factor and cytokine production from endothelial and SMCs. These early events lead to recruitment of leukocytes and eventually to SMC proliferation and migration, all of which act to form the mature atherosclerotic plaque. Of central importance is the understanding that hyperlipidemia is an inflammatory state. The connection between inflammation and hyperlipidemia is evident from the presence of foam cells, the hallmark of the fatty streak, which is the initial lesion of atherosclerosis. The foam cell is a macrophage named so because of the abundance of lipid within the cell. Macrophages bind and internalize modified lipoprotein particles via a number of “scavenger receptors,” including scavenger receptor-A family members, CD36, and macrosialin. Foam cells are able to further modify lipoproteins. In addition, lipoproteins can prove toxic to macrophages, leading to necrotic debris and free cholesterol clefts and ester within the lesion. This necrotic debris, along with the expression of the tissue factor and other molecules, leads to a very prothrombotic environment within the plaque and is a serious threat to local blood flow when there is loss of integrity of the barrier between the plaque and the blood stream.
Leukocyte Recruitment
Leukocytes, especially macrophages, play pivotal roles in atherosclerosis through their release of critical cytokines and growth factors that influence not only atherogenesis but also processes of plaque rupture and thrombosis. The process of leukocyte recruitment, attachment, and migration into the plaque is under the influence of a variety of molecules. As a response to injury, such as the accumulation of lipoproteins, endothelial cells express certain
adhesion molecules such as E-selectin, which interact with ligands on the surface of circulating leukocytes to begin a process of loose association and rolling along the surface of the vessel (4). Subsequent tight binding mediated by the integrin class of adhesion molecules stops the leukocytes prior to the process of diapedesis. Although their pathologic role is uncertain, soluble forms of cell adhesion molecules (CAMs) can be found in plasma. Human studies have demonstrated that plasma levels of intracellular CAMs (ICAM-1) and E-selectin correlate with clinical manifestations of coronary atherosclerosis (5).
adhesion molecules such as E-selectin, which interact with ligands on the surface of circulating leukocytes to begin a process of loose association and rolling along the surface of the vessel (4). Subsequent tight binding mediated by the integrin class of adhesion molecules stops the leukocytes prior to the process of diapedesis. Although their pathologic role is uncertain, soluble forms of cell adhesion molecules (CAMs) can be found in plasma. Human studies have demonstrated that plasma levels of intracellular CAMs (ICAM-1) and E-selectin correlate with clinical manifestations of coronary atherosclerosis (5).
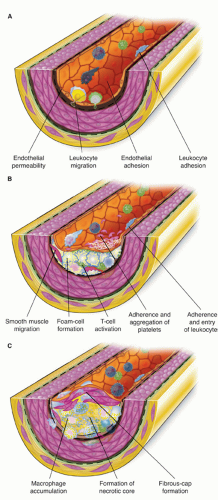 FIGURE 1-1 Stages of atherosclerotic plaque growth. A: Initial stage of atherosclerosis involves injury to the vessel wall with subsequent expression of inflammatory adhesion molecules, which leads to leukocyte recruitment. B: Intermediate lesions involve macrophages imbibing oxidized LDL leading to foam cell formation. There is continued leukocyte recruitment, formation of an early lipid core, and SMC proliferation and migration. C: The advanced or mature atherosclerotic plaque consists of a necrotic lipid core with foam cells, necrotic debris, and free cholesterol esters. In addition, there is a fibrous cap consisting of SMCs and extracellular matrix. (Adapted from: Ross R. N Engl J Med. 1999;340:115-126, with permission.) |
Also central to recruitment of leukocytes to areas of vascular injury, chemokines are a group of chemoattractant cytokines produced by a variety of somatic cells, including SMCs, endothelial cells, and leukocytes. One important chemokine of the C-C class, monocyte chemoattractant protein 1 (MCP-1), participates in the recruitment of monocytes in particular (6). Also critical is the C-X-C chemokine, interleukin-8 (IL-8), which participates in the recruitment of leukocytes to areas of vascular injury. IL-8 has been extensively documented in the recruitment of neutrophils (7), and more recent evidence suggests that the murine analogue of IL-8, KC, also plays a critical role in the recruitment of monocytes to injured areas (8).
As lesions mature, there tends to be excess accumulation of leukocytes at the “shoulder” regions of plaques, where the eccentric plaque merges with the more normal architecture of the vessel. This clustering is thought to make these shoulder regions more vulnerable to the consequences of atherosclerosis (9). In addition, it has long been observed that atherosclerotic lesions develop preferentially at areas of bifurcations within the coronary tree. This likely is related to disturbances in flow patterns and resultant areas of flow separation and altered shear stress, leading to preferential areas of upregulation of adhesion molecules and increased leukocyte recruitment (10). In addition, monocytes may contribute to vascular calcification in response to two cytokines: monocyte colony-stimulating factor and receptor activator of NF-kB (RANKL) (11). Emerging evidence indicates that atherosclerotic plaque calcification is positively correlated with vulnerability. Several inflammatory mediators have been shown to modulate arterial calcification, thus increasing the risk of plaque rupture. Among these factors, RANKL/OPG axis might be of particular interest as a promising biomarker of plaque vulnerability in subjects with diffuse coronary calcification (12).
Innate versus Adaptive Immunity
There are two major branches of the immune system: the innate or nonspecific arm and the adaptive or specific arm (13). There are several key differences that distinguish these two arms. The innate arm relies predominantly on phagocytic cells such as neutrophils and monocyte/macrophages, the cells most classically associated with atherosclerosis. The innate arm is not antigen-dependent and exhibits immediate response to foreign material. On the other hand, the adaptive arm is characterized by a specific antigen-dependent response that has the characteristic of conferring memory against the pathogen. Unlike the immediate response of the innate arm, the adaptive arm involves a lag time between exposure to the pathogen and response. The primary effector cells of this arm are lymphocytes. These two arms work in concert with dendritic cells of the innate immune system, representing a link between innate and adaptive immunity, as they are phagocytic cells, which then present antigens to cells of the adaptive system (14).
Innate Immune Response in Atherosclerosis
The monocyte is thought to be the first leukocyte recruited to the incipient atheroma after encountering complex signals that include soluble factors affecting general monocyte function in circulation. In addition, local factors affect the cells after monocyte adhesion to the endothelium and migration into the tissue. The defining cell of the fatty streak is the foam cell, a macrophage named so because of the abundance of lipid within the cell. Macrophages bind and internalize oxidized lipoprotein particles via a number of “scavenger receptors,” including scavenger receptor-A family members, CD36, and macrosialin. Foam cells can also further modify lipoproteins, making them more inflammatory. However, lipoproteins can also lead to toxicity of macrophages, leading to cell death and eventually leaving necrotic debris and free cholesterol clefts and ester within the lesion. Two main directions of monocyte-tomacrophage differentiation are recognized: Type 1, induced by inflammatory stimuli such as interferon (IFN)-g or lipopolysaccharide (LPS), and Type 2, induced by IL-4, IL-13, and other antiinflammatory cytokines. Type 1 macrophages (M1) produce high amounts of reactive oxygen species and inflammatory cytokines such as TNF (tumor necrosis factor) or IL-1b. Type 2 macrophages (M2) show high expression of scavenger receptors, produce extracellular matrix components and remodeling enzymes, secrete anti-inflammatory cytokines such as IL-1ra, CCL18, and IL-10, and express typical surface markers (15, 16 and 17). The phenotype of plaque-associated macrophages seems to be mixed, since infiltrating monocytes/macrophages are described to express markers of both M1 (TNF) and M2 (STAB1, CD163) (18). Macrophages amplify the inflammatory response through the secretion of cytokines such as TNF-a and IL 1-b (13). Other cells of the innate immune response that have been implicated in atherosclerosis include mast cells, natural killer cells, and neutrophils (19).
Adaptive Immune Response in Atherosclerosis
In contrast to the monocyte, the CD4+ T-cell is the primary cell of the adaptive arm of the immune response present at atherosclerotic sites. It is believed that these T-cells and their secreted cytokines influence progression and vulnerability of plaques (19). As an example, IFN-g appears to inhibit the growth of SMCs and promote apoptosis (programmed cell death) of these cells, leading to plaque vulnerability. In addition, IFN-γ appears to limit production of structural proteins (collagen and elastins) that SMCs secrete and may lead to a plaque more prone to rupture, as discussed below (20). CD4+ T-cells also express CD40 ligand on their surface, which is subsequently released as a soluble factor. Among its effects, soluble CD40 ligand appears to influence a variety of cell types to produce the highly procoagulant substance called tissue factor. There are subsets of CD4+ T-cells that have been identified by cellular receptors and the typical cytokines released from these cells. CD4+ TH1 T-cells are the predominant type at atherosclerotic lesions and are believed to promote atherosclerosis. CD4+ TH2 T-cells, on the other hand, are likely antiatherogenic. In addition, regulatory T-cells are present, which seem to suppress activation of other T-cells acting as an antiatherogenic mechanism.
Lastly, B-cells can also be identified within atherosclerotic lesions. Antibodies to oxidized LDL can be identified in human and animal models (19), and it is thought that this humoral immune response acts as a protectant against atherosclerosis, possibly by intercepting and neutralizing antigens before they reach sites of atherosclerosis. There are other evidences to suggest that infection with agents such as C. pneumoniae or perhaps viruses can create antibodies with autoimmune features that promote atherogenesis (21).
SMC Migration, Proliferation, and Extracellular Matrix Deposition
SMCs and their products are responsible for giving structure to the mature atherosclerotic plaque, which is, at first, little more than a collection of lipids and foam cells. Under the influence of growth factors and chemoattractants such as platelet-derived growth factor and thrombin, SMCs migrate out from the media into the neointima, where they begin to proliferate. In addition, SMCs produce extracellular matrix constituents, including collagen, proteoglycans, elastin, fibrinogen, fibronectin, and vitronectin. These proteins often account for a substantial volume of the plaque and are important in determining the structural integrity of the fibrous cap. Giachilli et al. have shown evidence that these cells express bone matrix proteins, which has been subsequently corroborated by other investigators (22, 23 and 24). This highlights the role of vascular smooth muscle cells in vascular calcification. In some patients, an additional process of mineralization of the atherosclerotic plaque will occur with deposition of calcium and osteopontin. Mineralization does not equate to a stable plaque, and has been associated with higher risk, especially in the elderly (25).
Plaque Angiogenesis and Hypoxia
A newly emerging area of interest is the potential role of angiogenesis in plaque growth and in the pathogenesis of atherosclerotic complications. New vasculature, under the influence of angiogenic growth factors such as hypoxia-inducible factor or vascular endothelial growth factor (26), may grow from the vasa vasorum within the adventitia into the plaque. These vessels may be disrupted and cause plaque hemorrhage independent of plaque rupture. The extravasated erythrocytes provide a local depot of cholesterol-rich redcell membrane and of heme, a source of iron, which is a stimulant for oxidative stress, which in turn would promote further growth.
In addition, analogous to tumor growth, these vessels may stimulate plaque growth. There is experimental evidence demonstrating inhibition of plaque growth by angiogenic inhibitors in a mouse model of atherosclerosis (27, 28). Furthermore, neo vessel density is higher in nonstenotic segments and stenotic noncalcified plaques than in normal segments or calcified lesions (29).
The concept of hypoxia not only promotes angiogensis but also contributes to proteolysis through the promotion of matrix metalloproteinases (MMPs), a family of interstitial collagenases that weaken the fibrous cap, and gellatinases capable of catabolizing nonfibrillar collagen, to which endothelial cells adhere (30, 31 and 32). Proteolysis would ultimately lead to dissolution of the plaque extracellular matrix, remodeling, and plaque vulnerability. Hypoxia promotes the formation of proinflammatory cytokines and leukotrienes, and activates Akt and β-catenin pathways with subsequent macrophage activation (31, 33). In addition, conditions of lipid accumulation in macrophages are amplified with accumulation of triglyceride containing cytosolic lipid droplets and adipose differentiation protein expression, even in the absence of exogenous lipids. The lipid accumulation is a result of increased triglyceride biosynthesis, reduced [beta]-oxidation of fatty acids, and increased expression of stearoyl-coenzyme A desaturase (SCD-1), an important enzyme in the synthesis of fatty acids (33, 34 and 35).
The Mature Atherosclerotic Plaque
The mature atherosclerotic plaque is therefore composed of several components, including a fibrous cap consisting of SMCs and extracellular matrix proteins overlying a necrotic lipid core consisting of free cholesterol esters, foam cells, other leukocytes such as T-cells, and necrotic debris of dead foam cells (Fig. 1-1). These plaques commonly are eccentric in nature, and there is heterogeneity in terms of the thickness of the cap as well as the distribution of leukocytes, which tend to cluster in shoulder regions. Both of these features have potential import in terms of propensity of plaques to cause ACS.
Vascular Remodeling
Although angiography remains the mainstay of diagnosis in coronary artery disease, its major limitation is that it provides information only on luminal encroachment of lesions, not on architecture of the vessel wall. Use of intravascular ultrasound has provided a much broader understanding of the nature of atherosclerosis by allowing systematic investigation of plaque architecture not only at sites of flow-obstructing lesions but throughout the vessel. Although the interventional cardiologist is most concerned with focal obstructive lesions in proximal portions of the vessel, it is important to realize that it is now recognized that atherosclerosis is almost always universally present throughout the coronary tree. The extent of impingement of the plaque on the lumen is controlled not only by the growth of plaque volume but also by vascular remodeling. Vascular remodeling involves restructuring of cellular or noncellular components of the wall, and can occur under a variety of stimuli (36




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
