Antiarrhythmic Drugs
Dan M. Roden
Overview
Drugs used for treating cardiac arrhythmias may not be completely effective and carry substantial risks for adverse effects. These adverse effects can be grouped into three broad categories: proarrhythmia, other cardiovascular effects such as bradycardia or heart failure, and noncardiovascular effects. Despite these shortcomings, antiarrhythmic drugs retain a place in the treatment of cardiac arrhythmias. A solid understanding of the mechanisms that underlie these adverse effects and of the drugs’ pharmacokinetics can help to minimize the likelihood of adverse effects during long-term treatment.
Historical Perspective
Treatment of cardiac arrhythmias has moved from the completely empiric administration of drugs to alleviate symptoms in the mid-eighteenth century to an increasing appreciation of the diverse mechanisms that may underlie cardiac arrhythmias and of the molecular mechanisms of action of older as well as newly available compounds. We now recognize that block of the function of membrane proteins, primarily ion channels and receptors, is the major mechanism underlying both the efficacy and toxicity of available antiarrhythmic drugs. However, with rare exceptions (notably the genetic ion channelopathies; see Chapter 96), dysfunction of these drug targets is not the mechanism responsible for most arrhythmias. It seems entirely likely that as our understanding of arrhythmia mechanisms continues to expand, new compounds devoid of serious toxicity that better target specific arrhythmic mechanisms may well be developed, thereby supplanting the highly nonspecific and poorly tolerated drugs currently available.
The increasing sophistication in understanding mechanisms underlying arrhythmias and antiarrhythmic drug actions and in nonpharmacologic approaches to arrhythmia management has resulted in more focused and narrower indications for antiarrhythmic drug therapy. Antiarrhythmic drugs continue to be first-line therapy for the acute suppression of ongoing arrhythmias. They are also used to prevent recurrence of chronic arrhythmias in patients in whom ablation has not been attempted, is not feasible, or has been unsuccessful. Although patients at high risk for serious arrhythmias can be identified, multiple trials have shown a detrimental effect of primary prophylactic antiarrhythmic drug therapy in such patients. In patients with implantable cardioverter-defibrillators (ICDs), antiarrhythmic drugs are sometimes required as adjunctive therapy to prevent arrhythmias that generate device discharges (see Chapter 76).
Trials of Pharmacologic Therapy to Prevent Sudden Cardiac Death
Sudden cardiac death (SCD) resulting from ventricular fibrillation (VF) accounts for about 25% of all deaths in adults in the United States (see Chapter 67). The recent history of antiarrhythmic drug development is largely focused on the issue of whether these agents alter SCD risk; this applies to settings in which they are used for symptomatic arrhythmias such as atrial fibrillation (AF), as well as to studies directly assessing their effects in patients at risk of SCD.
Large trials of β-blockers starting in the 1970s showed their value in reducing SCD incidence (1,2,3). Although calcium channel blockers exhibit antiarrhythmic effects in animal models, especially during myocardial ischemia (4), these drugs have produced either no change or an increase in SCD incidence in clinical trials (5,6). In the 1970s and 1980s, nonsustained ventricular arrhythmias were identified as a risk factor for SCD in patients convalescing from acute myocardial infarction (MI). Accordingly, the Cardiac Arrhythmia Suppression Trial (CAST), the first double-blind, randomized, placebo-controlled, well-powered study of antiarrhythmic drugs, was designed to test the hypothesis that suppression of
nonsustained ventricular arrhythmias in patients convalescing from acute MI would reduce mortality. However, CAST and other studies of sodium channel blockers showed that therapy with these agents unexpectedly increased mortality (7,8,9,10).
nonsustained ventricular arrhythmias in patients convalescing from acute MI would reduce mortality. However, CAST and other studies of sodium channel blockers showed that therapy with these agents unexpectedly increased mortality (7,8,9,10).
As a result, drug development moved rapidly away from sodium channel blockade and toward action potential (AP) prolongation as an antiarrhythmic intervention. This approach has theoretic advantages, such as preventing ischemic VF, lowering defibrillation energy requirements, and improving cardiac contractility (11,12). However, virtually all available AP-prolonging drugs act by blocking a specific cardiac potassium current, termed IKr; a class action of IKr blockers is the occasional development of marked QT prolongation and the potentially fatal polymorphic ventricular tachycardia (VT) called torsade de pointes (13). Mutations in the gene that encodes the protein responsible for IKr are one cause of the congenital long QT syndrome (LQTS), also associated with torsade de pointes (14,15). Trials of d-sotalol and dofetilide, IKr blockers devoid of other pharmacologic actions, showed no effect on or an increase in mortality (16,17,18). Taken together, these data suggest that if AP prolongation is to be successful as an antiarrhythmic mode of drug action, IKr blockade may not be the safest means of achieving this goal.
The most widely used antiarrhythmic, amiodarone, was originally developed as a thyroid hormone analog antianginal agent, and only subsequently was it found to exert antiarrhythmic effects (19,20). Although the drug has many pharmacologic properties including antiadrenergic effects that may explain its antiarrhythmic actions, the mechanisms by which it suppresses arrhythmias are not known. Multiple trials have assessed the effects of amiodarone to prevent SCD in patients at risk (21,22,23,24,25). Overall, there may be a small potential for benefit after MI (26,27), but probably not in heart failure (22,25); because the benefit is small (at most), and because the drug also has the potential for serious toxicity during long-term administration, it is not generally used for primary prophylaxis of SCD.
This chapter reviews antiarrhythmic drugs, traditionally defined as agents primarily developed or used for the therapy of arrhythmias. Recent studies with other classes of drugs widely used in cardiovascular therapy, notably hepatic 3-methylglutaryl coenzyme A (HMG CoA) reductase inhibitors (“statins”) and agents inhibiting the effects of activating the renin-angiotensin system (angiotensin-converting enzyme inhibitors and receptor blockers [ACE inhibitors and ARBs]) have demonstrated antiarrhythmic actions. The reduction in SCD by these agents may relate to their effects on vascular disease and so are perhaps unsurprising (28,29). However, there is increasing evidence that these agents also can reduce the incidence of AF (30,31,32,33,34). The mechanisms are not well understood, and antifibrotic, antiinflammatory, and antioxidant stress mechanisms have been invoked (35,36,37,38,39). These agents are not sufficiently well studied in arrhythmias to suggest that they be used in primary therapy, but they may be especially desirable in certain clinical settings; an example would be AF in hypertension, in which an ACE inhibitor would be a logical drug. Most importantly, the identification of such unanticipated antiarrhythmic actions points to heretofore unappreciated molecular pathways leading to arrhythmias and thus perhaps to newer therapies better targeted to underlying mechanisms.
Pathophysiology
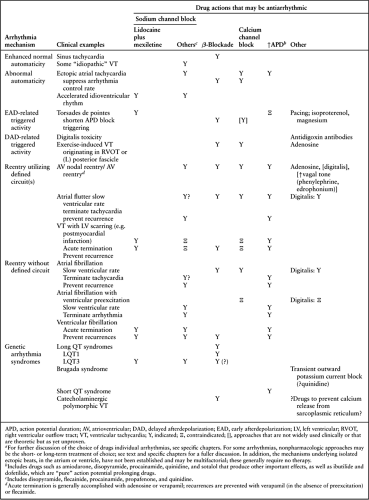
The three major mechanisms that underlie cardiac arrhythmias—enhanced automaticity, triggered activity, and reentry—are discussed in detail elsewhere (see Chapter 57). In most cases, choosing an antiarrhythmic drug for terminating ongoing arrhythmias or for suppressing recurrent episodes is based not on an understanding of specific mechanisms but rather on clinical experience. This experience includes not only determining what drug is likely to be effective against a particular arrhythmia but also estimating the risk for serious cardiac side effects from a particular drug in a particular patient, as well as estimating the risk for noncardiac side effects. This approach has proved effective because the drugs that are available are quite nonspecific (i.e., they produce a multiplicity of cardiac and noncardiac effects), and detailed characterization of the mechanisms underlying individual arrhythmic syndromes has not been considered part of routine clinical care until recently. Increasingly, precise characterization of mechanisms in individual arrhythmia syndromes may rationalize selection of antiarrhythmic drugs, to target specific “weak links” in those mechanisms, thus restoring normal rhythm. This approach, popularized as the Sicilian Gambit in the early 1990s (40), seems self-evident (Table 72.1). However, for many arrhythmias, there is insufficient information detailing the mechanisms, or in other cases appropriate drugs are not yet available to target weak links in individual arrhythmias. Conversely, an understanding of the putative mechanism underlying an individual arrhythmia in a particular patient may help the clinician to avoid using drugs that are likely to prove ineffective or to exacerbate an individual arrhythmia. Table 72.1 lists the mechanisms of the specific arrhythmias and provides clinical examples of arrhythmias caused by those mechanisms as well as drug effects that may be antiarrhythmic (or are contraindicated) in those mechanisms.
An especially appealing model for this approach is the way in which an emerging understanding of molecular mechanisms in the congenital arrhythmia syndromes has enabled identification of specific pharmacologic therapy for certain subtypes. Examples (see also Chapters 95 and 96) include variable efficacy of β-blockers as a function of molecular subtype in the congenital LQTSs (41), mexiletine or flecainide for the sodium channel–linked form of congenital LQTS (42,43), quinidine for the Brugada syndrome (44,45) and the short QT syndrome (46), and β-blockers for catecholaminergic polymorphic VT. In fact, in catecholaminergic polymorphic VT, experimental studies have identified calcium release from the sarcoplasmic reticulum as a major arrhythmogenic mechanism, and so drug therapy to prevent this arrhythmogenic phenomenon, for this rare syndrome and perhaps for more common diseases, may be a reasonable new approach (47).
Basic electrophysiologists have identified multiple ion currents in individual cardiac cells. This information allows identification not only of specific currents that are blocked by the drugs described in this chapter but also of factors such as rate and hypokalemia that can modulate this block. As described in this chapter, these data have direct implications for the clinical use of antiarrhythmic drugs. More recently, a combination of cellular electrophysiologic and molecular biologic techniques has enabled the cloning of individual genes whose protein products are responsible for individual ion currents, as well as studies of their function. Although this information could prove useful in designing new antiarrhythmic molecules, there are virtually no data attesting to the long-term safety of any drug whose primary mode of action is to block an ion channel. Thus, another implication of such advanced molecular information is that entirely new targets may be identified at which new drugs may act to modify cardiac electrophysiology in clinically effective and safe manners.
Clinical Profiles
Schemes that classify antiarrhythmic drugs by their major ionic mechanisms of action are commonly used. Their usefulness lies in that they may allow clinicians to predict the therapeutic
outcome of one drug based on the outcome of another. However, as mentioned previously, a major limitation of many currently available drugs is the likelihood of noncardiac side effects, which do not track with classification schemes. Moreover, members of the same drug class or subclass may exert subtle, or sometimes not so subtle, differences in pharmacologic actions, thus leading to different outcomes.
outcome of one drug based on the outcome of another. However, as mentioned previously, a major limitation of many currently available drugs is the likelihood of noncardiac side effects, which do not track with classification schemes. Moreover, members of the same drug class or subclass may exert subtle, or sometimes not so subtle, differences in pharmacologic actions, thus leading to different outcomes.
 |
Sodium Channel Blockade
Although many antiarrhythmic drugs block cardiac sodium channels (Table 72.2), a class I action, their clinical electrophysiologic effects vary. A major factor that underlies this variability is the rate at which a drug dissociates from the sodium channel (48). During each AP, drug associates with the channel, and with each diastolic period, drug dissociates. If this dissociation rate is rapid, such as a time constant (τ) less than 1 second, no channels are blocked with the next AP (Fig. 72.1). Thus, substantial sodium channel blockade occurs only with a rapidly dissociating drug such as lidocaine (class Ib) at fast rates (shorter diastole) or under conditions in which dissociation is slowed (e.g., ischemia). Conversely, with flecainide (class Ic), dissociation is so slow that substantial sodium channel blockade occurs even at slow rates. If sodium channel blockade accumulates (with lidocaine, at fast rates; with flecainide, at any rate), the most important electrophysiologic effect in cardiac tissue is to slow conduction; this is manifested by widening of the QRS interval. Thus, drug-induced sodium channel blockade is greater at fast rates, and hence QRS prolongation is exaggerated at more rapid rates. With rapidly dissociating drugs, such as lidocaine, no widening of the QRS interval manifests at usual rates. With other drugs, such as flecainide or quinidine, QRS prolongation is the rule at therapeutic dosages. Conduction slowing is actually a proarrhythmic effect in reentrant arrhythmias (see Chapter 57). The mechanisms by which sodium channel blockade suppresses arrhythmias are not completely understood but may include a bidirectional block in reentrant circuits (by effects on impulse propagation or on refractoriness) or suppression of abnormal automaticity. Sodium channel blockade can also produce clinically important increases in pacing and defibrillation energy requirements.
TABLE 72.2 Major Pharmacologic Effects of Antiarrhythmic Drugs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Proarrhythmic Effects of Sodium Channel Blockers
Therapy using sodium channel blockers such as flecainide has been associated with suppression of nonsustained ventricular
arrhythmias and of paroxysmal AF. However, conduction slowing in patients with known atrial flutter and in some patients with AF and no history of atrial flutter may sometimes lead to a marked slowing, but not termination, of the atrial flutter rate. With sufficient slowing, 1:1 AV conduction may ensue. To complicate the clinical picture further, because drugs such as flecainide block sodium channels more extensively at rapid than at slow rates, the ventricular complexes broaden and occasionally may even become aberrant. Thus, the clinical picture of this form of arrhythmia exacerbation is a patient with AF in whom wide, complex regular tachycardia then develops; it is therefore not a surprise that this arrhythmia is frequently misdiagnosed as VT (49,50). Management of this arrhythmia consists of recognizing the possibility of its occurrence, withdrawing the potentially offending antiarrhythmic agent, and using AV nodal blocking drugs both to establish the diagnosis as well as to control the ventricular rate. Adenosine may be useful in establishing the diagnosis.
arrhythmias and of paroxysmal AF. However, conduction slowing in patients with known atrial flutter and in some patients with AF and no history of atrial flutter may sometimes lead to a marked slowing, but not termination, of the atrial flutter rate. With sufficient slowing, 1:1 AV conduction may ensue. To complicate the clinical picture further, because drugs such as flecainide block sodium channels more extensively at rapid than at slow rates, the ventricular complexes broaden and occasionally may even become aberrant. Thus, the clinical picture of this form of arrhythmia exacerbation is a patient with AF in whom wide, complex regular tachycardia then develops; it is therefore not a surprise that this arrhythmia is frequently misdiagnosed as VT (49,50). Management of this arrhythmia consists of recognizing the possibility of its occurrence, withdrawing the potentially offending antiarrhythmic agent, and using AV nodal blocking drugs both to establish the diagnosis as well as to control the ventricular rate. Adenosine may be useful in establishing the diagnosis.
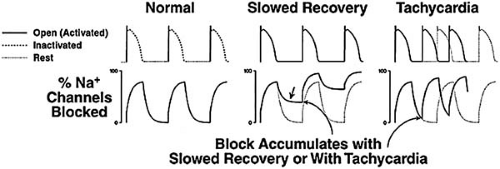 FIGURE 72.1. Rate-dependent sodium channel blockade. Left: A series of action potentials (APs), along with extent of sodium channel blockade (bottom). The sodium channel shuttles among three states during the AP: open, inactivated, and rest, as indicated in the top APs. Most sodium channel blockers that are used clinically bind to open and/or inactivated channels. Therefore, sodium channel blockade increases during the AP and decreases during each diastolic (rest) interval. A rapidly dissociating drug, such as lidocaine, is shown here. If the dissociation rate of the drug from the channel is decreased (middle), the extent of drug blockade can increase even in the absence of a change in rate. This is the case with lidocaine during ischemia or with flecainide. Right: Even with a rapidly dissociating drug such as lidocaine, sodium channel blockade can develop if the rate is accelerated. In the middle and right panels, the dotted lines indicate the data presented in the left panel. (From Roden DM, Echt DS, Murray KT, Lee JT. Clinical pharmacology of antiarrhythmic agents. In: Josephson ME, ed. Sudden cardiac death. Oxford: Blackwell Scientific, 1993:163–210. ) |
Conduction slowing in the ventricle usually only results in QRS prolongation. However, when a reentrant circuit caused by ventricular scarring is present (e.g., in the post-MI patient), conduction slowing by sodium channel blockers can increase the frequency of VT while often slowing its rate. Such drug-facilitated VT may be very resistant to cardioversion and may prove fatal (51). Some animal and a limited number of human studies (52,53) have suggested that sodium boluses may be effective in this setting.
Another proarrhythmic effect of sodium channel blockers is the increase in mortality observed in CAST. The mechanism responsible for this finding remains unclear. However, evidence from animal studies and the CAST database (54,55,56,57) strongly suggests that in the presence of sodium channel blockade, recurrent myocardial ischemia may enhance the likelihood of fatal ventricular arrhythmias such as VF. In CAST, the relative risk of sodium channel blockers in patients with transmural MI was 1.7 compared with those given placebo. However, among those patients with non–Q-wave MIs, a group thought to be at much higher risk for subsequent ischemic events, the relative risk was much higher, 8.7 (56).
Action Potential Prolongation
The hallmark of AP prolongation, a so-called class III effect, is QT prolongation; the designation class Ia has been used to refer to drugs that both block sodium channels and prolong QT interval. As mentioned earlier, drugs that prolong QT share a number of desirable features, including decreased defibrillation energy requirement, efficacy in models of ischemic VF, and increased contractility. Their major antiarrhythmic action is probably attributable to the prolongation of refractoriness that accompanies increased AP; this, in turn, is thought to suppress reentrant arrhythmias by decreasing the likelihood of unidirectional block (see Chapter 57).
Mechanisms of QT Prolongation Drug-Induced Torsade de Pointes
Torsade de pointes resulting from QT-prolonging antiarrhythmic drugs can occur in 1% to 5% of patients (58,59,60,61), with the exception of amiodarone, in which this form of proarrhythmia is rare (62,63). Risk factors include hypokalemia, hypomagnesemia, female gender, bradycardia, left ventricular hypertrophy, congestive heart failure (CHF), recent conversion from AF, and subclinical congenital LQTS (64,65); a short-long-short series of cycle-length changes usually occurs at the initiation of an episode. Multiple in vitro electrophysiologic changes together are thought to produce torsade de pointes. First, APs are prolonged by decreased outward current, usually IKr




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
