Tumor development is a multistep and complex process, requiring the transformation of a normal cell into a tumor cell through the accumulation of genetic alterations and the expansion of cell populations that have acquired growth and invasion advantages. However, in addition to the genetic and epigenetic alterations of transformation, another discrete process is required to allow tumor growth and metastasis—the induction of tumor vasculature known as the “angiogenic switch” (see also Chapter 8). Small tumors with a diameter <2 mm are dormant, receiving oxygen and nutrients by diffusion. Malignant growth and metastatic progression requires the tumor to develop an independent capillary network through two distinct processes, vasculogenesis and angiogenesis.1 In vasculogenesis, new blood vessels are formed when endothelial precursor cells (angioblasts) migrate and differentiate in response to stimuli such as growth factors. These nascent vascular trees are then remodeled and extended through angiogenesis. During tumor growth, circulating endothelial progenitor cells (derivatives of stem cells) can contribute to neovascularization.2 In contrast to vasculogenesis, angiogenesis is the formation of new blood vessels from preexisting vasculature.
Like many other physiological processes, angiogenesis is tightly orchestrated, regulated by a dynamic balance of proangiogenic and antiangiogenic molecules that drive and inhibit angiogenesis, respectively. The primary factor controlling angiogenesis is a lack of oxygen (hypoxia). Low oxygen tension triggers the secretion of proangiogenic factors and stimulates new vessel formation to increase oxygen supply. Tumor progression relies on tipping the balance in favor of molecules that drive angiogenesis. To achieve this, tumors undergo an angiogenic switch, creating an imbalance between the proangiogenic and antiangiogenic factors, resulting in increased angiogenesis and subsequent tumor progression and metastasis.3,4 The timing of this angiogenic switch during tumor progression is influenced by various factors including tumor type and environment.
Angiogenesis in tumors is aberrant, and tumor blood vessels have multiple structural and functional abnormalities. They are unusually dynamic and naturally undergo sprouting, proliferation, remodeling, or regression. The vessels are irregularly shaped and lack the normal architectural arrangement of arterioles, capillaries, and venules. Endothelial cells in tumors have abnormalities in gene expression, require growth factors for survival, and have defective barrier function to plasma proteins. Pericytes are relatively undifferentiated cells associated with the walls of physiologically normal small blood vessels and are also important elements of the vascular support structure of tumors, regulating endothelial cell survival and directing capillary growth.5 Pericytes on tumor vessels are also abnormal and, together with aberrant endothelial cells, generate a defective basement membrane. The effects of agents that inhibit factors involved in angiogenesis include stopping the growth of tumor vasculature, modifying existing vessels, and normalizing surviving vessels,6 indicating that tumor vasculature is reliant on continued expression of these factors for growth and survival.
Proangiogenic molecules that promote proliferation and migration are mainly receptor tyrosine kinase ligands, such as vascular endothelial growth factor (VEGF), fibroblast growth factors, platelet-derived growth factor (PDGF), and epidermal growth factor, but can also be of very different origin, such as lysophosphatic acid. Antiangiogenic molecules include statins such as angiostatin (a fragment of plasminogen that binds adenosine triphosphate [ATP] synthase and annexin II), endostatin, tumstatin, and canstatin (fragments of collagens that bind to integrins). Table 48.1 lists the known proangiogenic and antiangiogenic molecules. Changes in the balance of these proangiogenic and antiangiogenic molecules mediate the angiogenic switch.4 The precise role of many of these factors remains unclear; it is likely that ongoing and future research will attempt to further define the roles of these factors.
TABLE 48.1 List of Known Proangiogenic and Antiangiogenic Molecules and Agents
List of Known Angiogenic Growth Factors
List of Known Angiogenesis Inhibitors in the Body
Angiogenin
Angioarrestin
Angiopoietin-1
Angiostatin (plasminogen fragment)
Del-1
Antiangiogenic antithrombin III
Fibroblast growth factors: acidic (aFGF) and basic (bFGF)
From Understanding angiogenesis. Angiogenesis Foundation Web site. http://www.angio.org/understanding/understanding.html.
Angiogenesis does occur normally in the human body at specific times in development and growth; for example, it is an integral part of fetal development in utero. However, angiogenesis has a limited physiological role in healthy adults, where its functions are restricted to wound healing and the female menstrual cycle for a few days each month as new blood vessels form in the lining of the uterus. Inhibiting angiogenesis, therefore, has minimal adverse effects on normal physiological processes. As angiogenesis is essential for tumor growth, it is a rational therapeutic target. Agents targeting the effects of VEGF, one of the most important proangiogenic factors, and its downstream signaling pathways have been developed for the first-line treatment of patients with advanced non-small cell lung cancer (NSCLC) as well as other solid tumors, including metastatic colorectal, breast, and renal cell carcinoma.
VEGF: THE KEY MEDIATOR OF ANGIOGENESIS
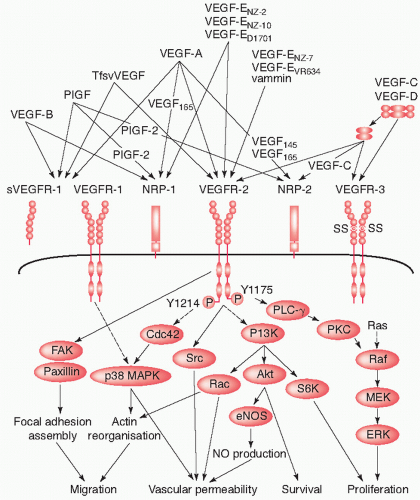
VEGF directly stimulates angiogenesis and is a key protein for sustaining tumor growth. VEGF is first synthesized inside tumor cells and then secreted into the surrounding tissue. When the VEGF ligand encounters endothelial cells, it binds to its main cell surface receptor, VEGF receptor 2. Ligand-receptor binding activates the endothelial cell and sets in motion a cascade of events that lead to the creation of new blood vessels (Fig. 48.1).7 First, the activated endothelial cell produces matrix metalloproteinases, a special class of degradative enzymes. These enzymes are released from the endothelial cell into the surrounding tissue where they degrade the extracellular matrix, allowing the migration of endothelial cells. As they migrate into the surrounding tissues, activated endothelial cells begin to divide, organize into hollow tubes, and gradually evolve into a mature network of blood vessels.8,9
VEGF and the VEGF signaling pathway have been the focus of an increasing number of studies as cancer targets. VEGF is expressed by many solid tumors, including melanoma,10 gastrointestinal,11 breast,12 central nervous system (CNS),13 ovarian,14 cervical,15 lung,16 hepatic,17 head and neck,18 and kidney19 carcinomas. Analysis of microvessel density can provide an indirect measure of angiogenesis. In many studies, VEGF levels have been shown to correlate with microvessel density, which is higher in advanced tumors compared with early stage tumors.20,21,22,23,24 A study of 105 patients with NSCLC demonstrated a significant association between VEGF expression and new vessel formation (p <0.0001), overall survival (p = 0.00003), and disease-free survival (p = 0.0004).21 In another study of specimens from 223 patients with operable NSCLC, 46.6% of cases had high VEGF expression. VEGF positivity was associated with high vascular grade disease (p = 0.009) and was prognostic for poor survival (p = 0.02).20 Several other studies have now confirmed a strong association between VEGF positivity and tumor grade and prognosis in NSCLC: Prognosis for patients with VEGF- positive tumors has been consistently shown to be significantly worse than that for patients with VEGF-negative tumors (p = 0.00322 and p = 0.01923).
VEGF exists in at least seven isoforms that result from alternative patterns of splicing of VEGF mRNA.8,9 One study evaluating the correlation between the expression of four different VEGF mRNA isoforms suggests that expression of VEGF189 mRNA isoform shows a greater correlation with survival and postoperative relapse time than expression of VEGF121, VEGF165, and VEGF206 mRNA isoforms.24 High VEGF189 mRNA isoform expression was associated with short survival (<24 months; p = 0.001) and early postoperative relapse (<12 months; p = 0.001), whereas no correlation was seen with VEGF165 and VEGF206, suggesting that only expression of certain VEGF isoforms may be prognostic indicators for NSCLC.24
Structurally, VEGF (sometimes referred to as VEGF-A or vascular permeability factor25) belongs to the platelet-derived growth factor (PDGF) family of cystine-knot growth factors.8 Other closely related proteins have been discovered (placenta growth factor [PlGF], VEGF-B, VEGF-C, and VEGF-D), and together these comprise the VEGF subfamily of growth factors.8 Several VEGF-related proteins are produced by viruses (VEGF-E) and in the venom of some snakes (VEGF-F).
VEGF growth factor ligands have specific VEGF receptors to which they bind to produce their physiological effects. VEGF-A binds to both VEGF receptor 1 (Flt-1) and 2 (KDR/Flk-1), but VEGF receptor 2 is believed to mediate almost all known cellular responses to VEGF.8 The function of VEGF receptor 1 is less well defined, although it is thought to modify VEGF receptor 2 signaling.8 Another function of VEGF receptor 1 may be to act as a decoy receptor, sequestering VEGF from VEGF receptor 2 binding, and this may be particularly important during embryonic vasculogenesis.26 A third receptor has been discovered (VEGF receptor 3), but VEGF-A is not a ligand for this receptor. VEGF receptor 3 mediates lymphangiogenesis in response to VEGF-C and VEGF-D binding.8,9,16
The biological effects of VEGF are thus initialized primarily through binding to VEGF receptor 2,27 which is expressed predominantly on vascular endothelial cells.28 VEGF receptor 2 is similar in structure to other tyrosine kinase receptors. It consists of seven extracellular immunoglobulin-like domains, a transmembrane region, and an intracellular domain with tyrosine kinase activity.29,30 VEGF binding to VEGF receptor 2 and subsequent receptor homodimerization are essential for stimulation of VEGF receptor 2-induced intracellular signaling, which is in turn essential to the VEGF signaling pathway.
MECHANISM OF ACTION OF ANTIANGIOGENIC AGENTS
Neutralizing the biological activity of VEGF reduces tumor vascularization and consequently inhibits tumor growth.6,31 A nonspecific tyrosine kinase inhibitor of the VEGF receptor prevented migration of endothelial cells, blocked capillary-like tubule formation, and prevented tumor blood vessel formation.31 Inhibition also prevented the formation of lung metastases and slowed progression of tumor growth. Importantly, for a potential therapeutic target, VEGF receptor inhibition had minimal effects on established blood vessels or blood flow. These findings indicate that a potent therapeutic role for VEGF inhibitors may be to prevent the formation of new blood vessels. Inhibition of VEGF signaling by blocking VEGF receptor 2 also inhibits angiogenesis, tumor growth, and invasion. VEGF receptor 2 blockade causes vessel regression and normalization as well as stromal maturation, resulting in a reversion to a noninvasive tumor phenotype. Importantly, this study suggests a crucial role for the stromal microenvironment (see Chapter 50) in regulating tumor phenotype and thus maintained and continuous VEGF inhibition may be essential for prolonged tumor suppression, which, in the clinical setting, may translate to prolonged progression-free survival (PFS).32,33,34
“Ghosts” or “tracks” of basement membrane and accompanying pericytes left behind after endothelial cells degenerate may provide a “scaffold” for microvascular regrowth in the absence of inhibition of angiogenesis. Given the transient and reversible effects of VEGF inhibition on tumors, with revascularization documented within weeks of withdrawal of inhibition,32,33,34 eradication of pericytes and ghosts of basement membrane may augment and prolong VEGF-inhibitor activity by decreasing the potential for vascular regrowth.34 Preclinical data suggest that dual targeting of pericytes and endothelial cells may be a more effective antiangiogenic strategy than antiendothelial cell targeting alone.5
Antiangiogenic Approaches As discussed, the evidence for the central role of VEGF-regulated angiogenesis in tumor growth and progression has provided a strong rationale for the development of agents that exert their antitumor effects through inhibition of various stages of the VEGF pathway. As such, the majority of antiangiogenic approaches to date have focused on the inhibition of this key proangiogenic factor. Of these, the most promising approaches are monoclonal antibodies directed at VEGF ligand and small molecule tyrosine kinase inhibitors that block the VEGF receptor(s) (Fig. 48.2).
Anti-VEGF Antibodies
Bevacizumab Bevacizumab, developed from the murine antihuman antibody A4.6.1, is a monoclonal antibody with a high affinity for VEGF.35,36 A4.6.1 was shown to potently suppress neovascularization and tumor growth and was humanized by site-directed mutagenesis to facilitate therapeutic use. The recombinant humanized antibody, bevacizumab, was able to bind VEGF with similar affinity to that of the original murine antibody and inhibit VEGF-induced proliferation of endothelial cells in vitro and tumor growth in vivo with potency and efficacy similar to A4.6.1. Bevacizumab exerts its antiangiogenic effects by binding to free, circulating VEGF, thereby inhibiting the binding of VEGF to its receptors, preventing VEGF ligand-receptor downstream signaling.36 To date, first-line bevacizumab combined with standard platinum-based chemotherapy has demonstrated clinical activity in a phase II trial37 and two phase III trials in patients with NSCLC.38,39
In the phase II trial, bevacizumab in combination with carboplatin and paclitaxel improved overall response and time to progression in patients with untreated advanced or recurrent NSCLC.37 In this trial, 99 patients were randomized to carboplatin/paclitaxel every 3 weeks (n = 32) or carboplatin/paclitaxel with bevacizumab 7.5 mg/kg (n = 32) or 15 mg/kg (n = 35) every 3 weeks. Patients with progressive disease who had received chemotherapy alone went on to receive single-agent bevacizumab. The combination of bevacizumab 15 mg/kg with carboplatin/paclitaxel increased response rate (31.5% vs. 18.8%) and prolonged time to progression (median 7.4 vs. 4.2 months; p = 0.023) with a 46% reduction in the risk of progression during treatment. With bevacizumab 7.5 mg/kg, there was a modest increase in response rate (28.1% vs. 18.8%) but no difference in time to progression (median 4.3 vs. 3.2 months). One-year survival was 47% for patients (n = 19) receiving chemotherapy alone who progressed and went on to receive single-agent bevacizumab. Bleeding was the most prominent adverse event (AE), mainly evident as minor mucocutaneous hemorrhage and major hemoptysis. Severe pulmonary hemorrhage was observed in six patients (9.1%) and led to four fatalities. An exploratory analysis identified increased risk of bleeding in patients with squamous cell histology, tumor necrosis and cavitation, and disease location close to major blood vessels.40 Further analysis excluding patients with squamous histology suggested that both doses of bevacizumab increased response rate, time to progression, and survival compared with chemotherapy alone and with only a small increased risk of bleeding (∽4%).37
Based on the positive results of the phase II trial, a randomized phase III trial of bevacizumab-based first-line therapy was conducted by the Eastern Cooperative Oncology Group (ECOG).38 In the E4599 trial, 878 patients with recurrent or advanced NSCLC (stage IIIB or IV) were randomized to bevacizumab 15 mg/kg with carboplatin/paclitaxel (n = 434) or carboplatin/paclitaxel alone (n = 444) every 3 weeks for six cycles until disease progression or unacceptable toxicity.38 The 15 mg/kg dose was chosen based on its activity in the phase II trial and based on exploratory analyses from the phase II trial, patients with squamous histology were excluded from E4599. Patients with brain metastases or clinically significant hemoptysis were also excluded. The primary end point was overall survival.
A survival advantage was demonstrated for bevacizumab-based first-line therapy compared with conventional chemotherapy alone. Median overall survival was 12.3 months for bevacizumab-based therapy compared with 10.3 months for chemotherapy alone (hazard ratio [HR] = 0.79; p = 0.003). Median PFS was 6.2 and 4.5 months, respectively (HR = 0.66; p <0.001), with corresponding response rates of 35% and 15% (p <0.001). Exclusion of patients with squamous cell histology resulted in a reduction in the rates of clinically significant bleeding compared with the earlier phase II trial.37 Notably, the incidence of severe pulmonary hemorrhage in E4599 was reduced when compared with the patients with nonsquamous histology in the phase II trial (1.9% vs. 4%, respectively). There were 15 treatment-related deaths among patients receiving bevacizumab-based therapy, including five from pulmonary hemorrhage. VEGF levels before treatment were measured and found not to correlate with overall survival. An exploratory analysis found that despite improvements in PFS, there was no improvement in overall survival for women treated with bevacizumab-based therapy compared with chemotherapy alone (13.3 vs. 13.1 months). This was not true for men (11.7 vs. 8.7 months). The reasons for this are not clear.
In a pre-planned subset analysis of patients in E4599 with adenocarcinoma histology, bevacizumab-based therapy extended median overall survival to 14.2 months compared with 10.3 months for chemotherapy alone (HR = 0.69, a 31% reduction in the risk of death).41 A retrospective subset analysis of elderly patients (≥70 years of age), representing 26% of study patients, found a trend toward higher response rates (17% vs. 29%) and PFS (4.9 vs. 5.9 months) but no difference in overall survival (12.1 vs. 11.3 months) for paclitaxel/carboplatin versus paclitaxel/carboplatin plus bevacizumab, respectively. Paclitaxel/carboplatin plus bevacizumab was also associated with a significantly higher incidence of grade 3 to 5 toxicities than paclitaxel/carboplatin (61% vs. 87%; p <0.001).42
The improved outcome for bevacizumab-based first-line therapy in NSCLC has recently been confirmed in a second randomized phase III trial of bevacizumab in combination with cisplatin/gemcitabine. In the Avastin in lung (AVAiL; BO17704) trial, the combination of bevacizumab with cisplatin/gemcitabine improved PFS when compared with the platinum-based regimen alone.39 In this trial, 1043 patients with advanced or recurrent nonsquamous NSCLC were randomized to receive cisplatin/gemcitabine for up to six cycles plus bevacizumab 7.5 mg/kg (n = 345), bevacizumab 15 mg/kg (n = 351), or placebo (n = 347) every 3 weeks until disease progression or unacceptable toxicity. As in E4599, patients with squamous cell histology, baseline hemoptysis, or brain metastases were excluded to minimize toxicity. In this trial, patients with tumors in proximity to or abutting major vessels were also excluded. Following the positive survival results of the E4599 trial for bevacizumab-based therapy versus chemotherapy alone, the primary end point of AVAiL was amended from overall survival to PFS.
Both doses of bevacizumab-based therapy demonstrated a significant PFS benefit compared with chemotherapy alone. Median PFS for bevacizumab 7.5 mg/kg and 15 mg/kg versus placebo was 6.7 versus 6.1 months (HR = 0.75; p = 0.003) and 6.5 versus 6.1 months (HR = 0.82; p = 0.03), respectively. Objective response rates were 34.1%, 30.4%, and 20.1% for bevacizumab 7.5 mg/kg, 15 mg/kg, and placebo, respectively. The PFS benefit did not translate into a significant overall survival benefit, possibly due to the high use of efficacious second-line therapies in the trial. Median overall survival for bevacizumab 7.5 mg/kg and 15 mg/kg versus placebo was 13.6 versus 13.1 months (HR 0.93; p = 0.420) and 13.4 versus 13.1 months (HR 1.03; p = 0.761), respectively.43 The majority of AEs were grade 1 or 2 and the incidence of grade ≥3 AEs was similar across arms. Although 9% of patients received therapeutic anticoagulation, severe pulmonary hemorrhage rates were ≤1.5% for all arms, highlighting the role of patient selection in managing the risk of bleeding. The AE fatality rate (from any cause) was similar across treatment groups (4% to 5%). Although the AVAiL trial was not powered to compare the two bevacizumab doses directly, efficacy and safety data were similar for both doses. Table 48.2 presents a summary of results for the clinical trials of bevacizumab-based first-line therapy in NSCLC.
In addition to the two phase III trials, E4599 and AVAiL, the safety and clinical activity of bevacizumab-based therapy is being investigated in the Safety of Avastin in lung (SAiL; MO19390) trial. SAiL is a large (n = >2,100 patients are expected), open-label, multicentre, single-arm trial that is being conducted to generate further safety and efficacy data for bevacizumab combined with a range of standard first-line chemotherapy regimens in a broad ‘real-life’ clinical population of patients with advanced non-squamous NSCLC. The primary objective is to evaluate the safety profile of bevacizumab-based therapy. Interim results show that no new safety signals have been reported in this large study and that the safety profile of bevacizumab-based therapy is consistent with that reported in previous phase III trials.44 Interim results from SAiL further confirm the safety profile of bevacizumab when used in combination with a wide range of chemotherapies. Final data, including efficacy outcomes, are anticipated in 2010. The clinical potential and therapeutic advance offered by bevacizumab are being evaluated further in an ongoing clinical research program. The majority of these (approximately 80%) are phase II trials with most evaluating the potential benefit of the addition of bevacizumab to different chemotherapy regimens, including pemetrexed/oxaliplatin,45,46 pemetrexed/carboplatin,47,48,49 docetaxel/carboplatin,50 oxaliplatin/gemcitabine,51 and gemcitabine/carboplatin.52 However, many trials are combining bevacizumab not only with standard chemotherapy but with other biological therapies, most notably erlotinib in almost one quarter of trials listed. More creative approaches are also being studied, including novel combinations with targeted therapies and improved prognostic profiling to identify those patients most likely to benefit from specific therapeutic interventions. The increasing sophistication of clinical trials reflects the number of innovative agents available or in development, and our improved understanding of the underlying pathogenesis and tumorigenic processes that allow for rational combination of therapies with potentially complementary antitumor activity. Several of the significant ongoing bevacizumab trials, including trials in the neoadjuvant setting and trials in specific patient populations, deserve a special mention and are summarized later.
TABLE 48.2 Summary of Phase II and III Data for Bevacizumab-Based Therapy37,38,39,43
* p values significant (<0.05) relative to control for asterisked values.
N/A, not applicable; N/G, not given; ORR, overall response rate; OS, overall survival; PFS, progression-free survival.
Combining Bevacizumab with Other Targeted Agents The concept of using two novel agents, such as an epidermal growth factor receptor (EGFR) inhibitor and an anti-VEGF agent, is intriguing. As discussed, the angiogenesis pathway is crucial in lung cancer development. Data have also demonstrated the significant therapeutic involvement of the EGFR pathway in tumorigenesis.53 Therefore, inhibiting both pathways to exert a greater combined antitumor effect, but with fewer nonspecific toxicities than chemotherapy,54 represents a rational therapeutic approach.
Erlotinib (Tarceva), an oral HER-1 (human epidermal receptor 1)/EGFR tyrosine kinase inhibitor has shown a survival benefit in the treatment of lung cancer in phase III trials and is approved for the treatment of locally advanced or metastatic NSCLC in patients failing at least one prior chemotherapy regimen.55 A phase I/II trail of 40 patients with advanced, nonsquamous NSCLC who had failed at least one prior chemotherapy regimen demonstrated a 20% response rate, 6.2 months PFS, 12.6 months overall survival, and no grade III/IV toxicity when bevacizumab (15 mg/kg) was combined with erlotinib (150 mg daily).56 These results led to a randomized phase II trial comparing chemotherapy alone (docetaxel 75 mg/m2 or pemetrexed 500 mg/m2 every 3 weeks) to chemotherapy plus bevacizumab (15 mg/kg) or bevacizumab plus erlotinib (150 mg daily) in 120 patients with nonsquamous NSCLC who had progressed following platinum-based chemotherapy. Grade 3/4 toxicities were greater in the chemotherapy treatment arms with a higher proportion of patients discontinuing treatment caused by AEs (24% for chemotherapy alone vs. 28% for chemotherapy plus bevacizumab and 13% for erlotinib plus bevacizumab). The response rate was higher for the erlotinib plus bevacizumab combination (18%), versus 12% for chemotherapy alone and 13% for chemotherapy plus bevacizumab. Compared with chemotherapy alone, patients who received chemotherapy plus bevacizumab and erlotinib plus bevacizumab had superior outcomes, respectively, in terms of median PFS (3.0 vs. 4.8 vs. 4.4 months), overall survival (8.6 vs. 12.6 vs. 13.7 months), and 1-year survival (33% vs. 53.8% vs. 57.4%).57
Based on these results, the BeTa lung trial (OSI3364g/NCT00130728) has opened. This phase III trial will compare erlotinib with erlotinib plus bevacizumab in 655 patients with nonsquamous NSCLC who have progressed following standard first-line therapy. The primary end point is overall survival; secondary end points are PFS, response rate, and response duration. ATLAS (AVF3671g/NCT00257608) is another phase III trial assessing the efficacy and safety of maintenance bevacizumab with or without erlotinib following chemotherapy (carboplatin/paclitaxel, cisplatin/gemcitabine, or carboplatin/docetaxel) plus bevacizumab before randomization to bevacizumab with or without erlotinib, in 1150 previously untreated patients. The primary end point is PFS and secondary end points include safety of bevacizumab during the chemotherapy phase (by chemotherapy regimen) and safety of bevacizumab plus erlotinib versus bevacizumab plus placebo. Of note, patients with squamous cell histology and brain metastases are excluded from this trial.
Given that bevacizumab is administered until disease progression, a key issue to address is when it is appropriate to add erlotinib, which is approved in the second-or third-line settings. The value of earlier versus delayed initiation of erlotinib should be assessed, as should the optimal timing of bevacizumab/erlotinib therapy in the disease pathway.
A two-stage phase II trial evaluating up-front administration of erlotinib (150 mg daily) with bevacizumab (15 mg/kg every 21 days) is currently ongoing. Preliminary data from 33 patients indicate major toxicities (>10% of patients) to be rash and diarrhea. The primary end point of nonprogression at 6 weeks has been met in 75% of patients. With a median follow up of 6.3 months, the median time to progression is 5.5 months.58 More mature data with correlative studies are pending.
Only gold members can continue reading. Log In or Register to continue