Chapter 143
Aneurysms Caused by Connective Tissue Abnormalities
James H. Black III
The primary structural proteins of connective tissue are composed of collagen and elastin, which vary in type and amount within each of the body’s tissues; those constitutive of blood vessels are listed in Table 143-1. A connective tissue disease is a genetic disease in which the primary target is either collagen or elastin protein assembly, disruption of which leads to an inherent predisposition to degeneration, loss of structural integrity, and consequent aneurysm formation or spontaneous vascular dissection and rupture. Although inflammation may affect these proteins and induce structural damage in some patients, such conditions often imply some element of autoimmune disorder and are termed collagen vascular diseases or mixed connective tissue diseases. Such conditions and arteritides related to the vascular tree, which are considered in Chapters 78, 79, and 80, may have genetic profiles that predispose to their development. Although clustering of aneurysms in multiply affected family members within these arteritides may indicate some element of an inheritance pattern, there are often greatly varying levels of expression and penetrance, and no defined genetic test is available to assist treatment. For the sporadic occurrence of degenerative aneurysms within kindreds, the underlying mechanisms are considered in Chapter 129. Herein we seek to define the common connective tissue diseases affecting the arterial tree, which have a studied natural history, a defined basis for genetic inheritance, and sufficiently understood pathophysiologic mechanisms to guide treatment paradigms. These “heritable disorders of connective tissue”1 have severe vascular manifestations and most commonly include Marfan’s syndrome, the vascular type of Ehlers-Danlos syndrome (EDS IV), Loeys-Dietz syndrome (LDS), and familial thoracic aortic aneurysm and dissection (TAAD).
Table 143-1
Structural Elements of Blood Vessels
| Structural Proteins | Approximate Amount (% Dry Wt) | Function |
| Type I collagen | 20-40 | Fibrillar network |
| Type III collagen | 20-40 | Thin fibrils |
| Elastin, fibrillin | 20-40 | Elasticity |
| Type IV collagen, laminin | <5 | Basal lamina |
| Types V and VI collagen | <2 | Function unclear |
| Proteoglycans (>30 types) | <3 | Resiliency |
Marfan’s Syndrome
Antonin-Bernard Marfan, a Professor of Pediatrics in Paris, in 1896 encountered a 5-year-old girl with congenital deformation of all four limbs.2 By the time she was 11 years old, thoracolumbar kyphoscoliosis, pectus carinatum, and signs of tuberculosis had developed.3 She died at age 16 from infection, and no autopsy was performed to document any vascular involvement. The first description of aortic pathology in Marfan’s syndrome was published in 1943, a year after Marfan’s death.4 Although he correctly identified the many mendelian features of the condition that would eventually bear his name, the pleiotropic disorder has benefited from decades of further description of clinical manifestations, molecular pathogenesis, and emerging therapeutic options.
Epidemiology and Natural History
The incidence of Marfan’s syndrome is about 2 to 3 per 10,000 individuals, but this estimate relies on proper recognition of all affected and genetically predisposed individuals.5 A population-based study in Scotland found an incidence of 1 in 9802 live births,6 although this number would underestimate the true incidence inasmuch as the features of Marfan’s syndrome, particularly the skeletal ones, become more apparent with growth. Furthermore, even though the disorder is passed as a dominant mendelian trait, about 25% of cases are due to sporadic de novo mutations.7 The disease has no gender predisposition. Its incidence is increased in athletes, particularly in basketball and volleyball players, because the characteristic tall stature with long-bone overgrowth (dolichostenomelia) confers a competitive advantage. In a screening study of 415 high-school basketball and volleyball athletes performed with standard echocardiography, 4 (1%) of these subjects exhibited aortic root enlargement greater than 4.6 cm, and Marfan’s syndrome was diagnosed in 2.8
The life span of individuals with Marfan’s syndrome was significantly shortened before the widespread and successful refinement of aortic root surgery. Before adoption of thresholds for aortic root replacement, the cause of death was cardiovascular (aortic rupture, aortic dissection, or valvular disease) in 90% of cases.9 A report in the 1970s on the life expectancy of patients with Marfan’s syndrome described longevity as only two thirds that of unaffected individuals, with life-table mortality curves deviating in infancy.9 However, a later assessment of longevity in patients with Marfan’s syndrome describes a nearly normal life expectancy as a result of improvement and refinement in diagnosis and treatment, particularly of the cardiovascular manifestations of the disorder.10
Pathogenesis
As early as 1955 it was suggested that the basic structural defect in Marfan’s syndrome was localized to the elastic fiber,11 with skin and aorta from affected patients showing decreased elastin content and fragmentation of elastic fibers.12,13 Yet the elastin gene and molecule were a poor target to explain the clinical manifestations of Marfan’s syndrome in tissues that are devoid of elastin, such as bone and the ciliary zonules in the eye. Further histochemical analysis demonstrated that the amorphous fragmented elastin tissues were surrounded by a rodlike material with a distinct staining pattern and distinguishable susceptibility to enzymatic digestion.14,15 These so-called microfibrils are 10 to 14 nm in diameter and are constituents of all connective tissue.15 Sakai et al16 first identified fibrillin-1 (FBN1) as the principal component of the extracellular matrix microfibril, present in all tissues with the phenotypic manifestations of Marfan’s syndrome. Additional linkage analysis mapped the Marfan’s syndrome locus to 15q21.1.17 To date, all of the mutation analyses have identified the FBN1 gene as the sole locus for classic Marfan’s syndrome, and most families have unique or private mutations. More than 550 mutations have been reported within the FBN1 gene.18 Neither the location of the mutation nor the type of amino acid altered is sufficient to predict phenotype in any individual patient. An exception is mutations within exons 24 to 32, which are associated with a severe form of Marfan’s syndrome diagnosed in early childhood.19 The mutation is passed in an autosomal dominant manner with complete penetrance, so 50% of the offspring of an affected individual can inherit a genetic predisposition to the disorder.5
Role of Fibrillin
The FBN1 gene contains 65 exons spanning 235 kilobases of genomic DNA.20,21 The gene encodes a 350-kD glycoprotein that is highly conserved among different species, thus suggesting its critical homeostatic importance.21 Initial findings indicated that the defect in Marfan’s syndrome was caused by activity of the mutant protein on the deposition, stability, or function of the neighboring normal protein—the “dominant negative effect.” This would, of course, bode poorly for the development of productive treatment strategies because it implied that patients had an immutable structural predisposition to tissue failure later in life.5 However, the association between a mutant fibrillar protein exerting a dominant effect and severity of disease was refuted by the discovery of patients with severe disease and near absence of mutant protein because of a premature termination codon created by their specific mutation.22 To reconcile these disparate findings, murine models of Marfan’s syndrome were developed to provide the opportunity to study the earliest pathogenetic abnormalities in elastogenesis and aortic aneurysm formation.23,24 These models demonstrated that normal FBN1 molecules are not needed to assemble an elastic fiber; rather, microfibrils are required to maintain normal elastic fibers during postnatal life. If proper connections among elastic fibers and vascular smooth muscles cells are not suitably maintained, cells adopt matrix-degrading enzymes such as matrix metalloproteinase types 2 and 9. Thereafter, aortic wall homeostasis is perturbed, and inflammation as well as calcification and structural weakening of elastic fibers may ensue. This pathology has been observed in large muscular arteries from patients with Marfan’s syndrome,25 leading to appreciation of the classic lesion of cystic medial necrosis in large arteries of individuals with Marfan’s syndrome. Verhoeff–van Gieson staining of elastic fibers in the aorta demonstrates classic lamellar disorganization in Marfan’s syndrome secondary to errant elastic fiber maintenance (Fig. 143-1).
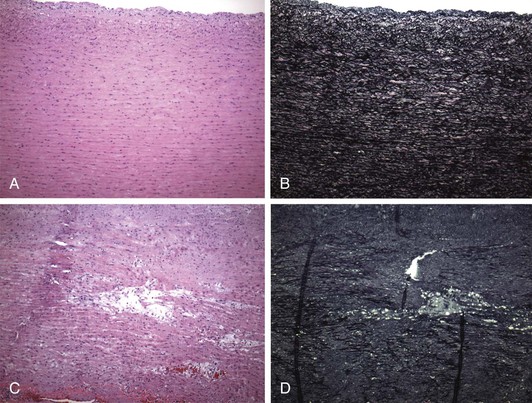
Figure 143-1 A and B, Photomicrographs illustrating the regular and parallel nature of the elastic lamellae found within the media of the normal ascending aorta. The lamellae are composed of elastic fibers running in parallel with intervening smooth muscle, ground substance, and collagen. The Verhoeff–van Gieson (VVG) stain highlights these major elastic fibers (black) (A, hematoxylin-eosin [H&E], ×100; B, VVG, ×100). C and D, Photomicrographs showing profound fragmentation of the elastic fibers, with spaces left within the media of the ascending aorta. Though often referred to as cystic medial degeneration, the spaces created by this fragmentation lack a lining and hence are not truly “cysts.” These spaces often contain increased amounts of glycoproteins. The VVG stain further highlights the severe elastic fiber (black) fragmentation. The vertical black lines are fixation artifacts from folding of the elastic sheet (C, H&E; D, VVG). (Plates courtesy Joseph Maleszewski, MD, Johns Hopkins Hospital Department of Pathology.)
Interaction with Transforming Growth Factor
The discovery of the role of microfibrils in regulating cytokines has further advanced our understanding of the pathogenesis of Marfan’s syndrome and raised the possibility of a new treatment paradigm. FBN1 shares a high degree of homology with the latent transforming growth factor-β (TGF-β) binding proteins. The TGF-β cytokines are secreted as large latent complexes consisting of TGF-β, a latency-associated peptide, and one of three latent TGF-β binding proteins (Fig. 143-2).26 In normal trafficking, the large latent complex is sequestered and bound to microfibrils, and TGF-β cytokine signaling is prevented. Once the mature cytokine is released from the binding proteins, interactions with cell surface receptors and downstream signaling can occur via the well-defined SMAD (nuclear proteins involved in signal transduction for TGF-β ligands) transcription factor pathways, which modulate the effects of TGF-β. The homology between fibrillins and latent TGF-β binding proteins prompted the hypothesis that microfibrils may play a role in the trafficking of TGF-β and its activation. Credence has been lent to this hypothesis with the demonstration of elevated TGF-β activity and free TGF-β levels in FBN1-deficient mice during developmental septation of the lung27 and the development of myxomatous cardiac valvular changes.28 As depicted in Figure 143-2, without proper microfibrils, the TGF-β complex cannot form, thereby leaving TGF-β in the milieu to incite excess signaling. This pathogenetic mechanism of dysregulated TGF-β activity seems more plausible in explaining the clinical features of Marfan’s syndrome that are poorly reconciled with structural failure, such as long-bone overgrowth, craniofacial abnormalities, and muscle hypoplasia.28
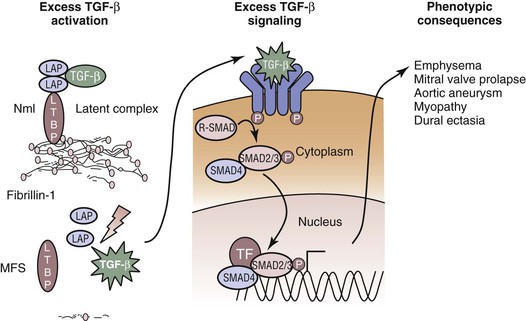
Figure 143-2 Excess activation of transforming growth factor-β (TGF-β) causes many of the features of Marfan’s syndrome. Normal TGF-β metabolism requires binding of the cytokine to several proteins, including microfibrils, to prevent excess signaling. In Marfan’s syndrome, lack of normal microfibrillar assembly allows TGF-β to remain unsequestered in the extracellular space. As a consequence, excess TGF-β signaling can occur on the cell surfaces of TGF-β receptors. Once the TGF-β binds to its receptor, downstream receptor-associated SMAD proteins translocate to the nucleus to modulate transcriptional activity, alter protein expression, and yield phenotypic change. LAP, Latency-associated peptide; LTBP, latent TGF-β binding protein; MFS, Marfan’s syndrome; Nml, normal; TF, tissue factor.
Clinical Manifestations and Diagnostic Evaluation
Marfan’s syndrome is a multisystem disorder with manifestations principally within the cardiovascular, ocular, and skeletal systems. The disorder occurs worldwide, with no gender or race predilection. However, the cardinal manifestation of aortic root aneurysm and its risk of life-threatening aortic dissection or rupture led to a shortened life expectancy. Consequently, the leading cause of mortality was cardiovascular in more than 90% of cases (aortic dissection, valve disease, or congestive heart failure), which decreased life expectancy to approximately two thirds that of unaffected individuals.5 Improvements in recognition of Marfan’s syndrome and surgical advances have returned life expectancy to the nearly normal range by addressing the most threatening manifestations of Marfan’s syndrome—aortic catastrophe in the form of dissection or rupture of the ascending aorta.
Diagnostic Criteria
Ghent Criteria.
Clinical diagnostic criteria for Marfan’s syndrome were outlined at the International Nosology of Heritable Connective Tissue Disorders in 1986 during the Connective Tissue Meeting in Berlin.29 Thereafter, the recognition that many individuals diagnosed by means of these original criteria did not have the FBN1 mutation (genetic testing became possible after 1986) led to a focused revision in 1996 and another in 2010.30,31 Termed the Ghent criteria, the current nosology places emphasis on the cardinal manifestations of the syndrome, namely aortic root aneurysm, ectopia lentis, and genetic testing, versus the less predictive skeletal findings (Box 143-1). Once Marfan’s syndrome is diagnosed in an individual, all first-degree relatives should be evaluated for the presence of the condition. In children, repeated evaluations may be required to avoid missing of the disorder in its evolution.
Genetic Testing.
The role of clinical genetic testing in establishing a diagnosis remains limited because more than 500 mutations have been found and 90% of the mutations are unique within a pedigree.18 Even within families in which the same mutation is shared, phenotypic variation is prominent. Thus, exacting a genotype-phenotype correlation is difficult.32 Furthermore, approximately 25% of patients with the disorder have a de novo mutation, thereby limiting the feasibility of a more focused analysis. In addition, it is estimated that 10% of the mutations in the FBN1 gene that cause Marfan’s syndrome are missed by conventional screening methods.33 At present, the diagnosis of Marfan’s syndrome rests primarily on physical clinical assessment based on the Ghent criteria.
Differential Diagnosis
Other conditions also associated with FBN1 mutations may be considered in the differential diagnosis of Marfan’s syndrome. The MASS phenotype is based on the association of mitral valve prolapse, myopia, mild aortic root dilatation, striae, and mild skeletal changes.34 The skeletal features of MASS often include the mild manifestations of tall stature, mild dolichostenomelia (long-bone growth), and scoliosis. Occasionally, a major Ghent criterion from the skeletal system may be met but no other major criteria are noted. For patients with MASS, mutations in the FBN1 gene have generally created premature termination codons, and the mutant transcript can be easily and rapidly degraded.35
Shprintzen-Goldberg syndrome is characterized by craniosynostosis, facial hypoplasia, anterior chest deformity, arachnodactyly (long, spider-like fingers), and aortic root dilatation. Developmental delay is also common. Point mutations in FBN1 have been found in some affected individuals, but phenotypic heterogeneity, specifically regarding development, retardation, and aortic root involvement, probably indicates substantial genotypic variation.36 The aortic root enlargement in Shprintzen-Goldberg syndrome is similar to that in Marfan’s syndrome.
Homocystinuria is caused by a deficiency of cystathionine β-synthase. Patients with homocystinuria often have tall stature, long-bone overgrowth, and ectopia lentis, but no aortic enlargement. The inheritance is autosomal recessive, and affected individuals often have mental retardation, a history of thromboembolism, and coronary artery disease. Plasma homocysteine values are typically markedly elevated, easily distinguishing this disease from Marfan’s syndrome.5
Congenital contractural arachnodactyly (CCA) shares many skeletal features with Marfan’s syndrome, without the ocular and cardiovascular manifestations. The mutation in the few patients reported in the literature is located in the FBN2 gene,37 and physical therapy is key to maintaining joint range of motion. It has been suggested that President Abraham Lincoln had Marfan’s syndrome, but pictures of his later years suggest large joint contractures, which are more typical of congenital contractural arachnodactyly.
The overlap of LDS and Marfan’s syndrome is considered later in this chapter.
Surveillance
Marfan’s syndrome is a pleiotropic disorder, and surveillance of the many systems at risk for abnormality is prudent. The cardinal manifestations are ocular, skeletal, and cardiovascular. Regular examinations by an ophthalmologist for slit-lamp testing, a cardiologist for imaging of the aortic root, and an orthopedist for the development of scoliosis should be performed on an annual basis. In this section, focus is placed on aortic and vascular pathology. For recommendations with regard to the other body systems, the reader may find useful information at the National Marfan Foundation website (www.marfan.org). The clinical manifestations within the cardiovascular system that require preventive attention involve the atrioventricular valves, the annuloaortic valve mechanism, and the aortic root and ascending aorta.
Valvular Disease
Thickening of the atrioventricular valves is very common in Marfan’s syndrome and often associated with mitral or tricuspid valve prolapse.5 In about 25% of patients, the mitral valve prolapse may progress to severe mitral regurgitation.38 Mitral regurgitation is the most common indication for cardiac surgery in infants and children with the disorder.38 In children with early onset of Marfan’s syndrome, congestive heart failure, pulmonary hypertension, and death may result from this mitral insufficiency, which is the leading cause of morbidity and mortality in young children with the disorder.38 Interestingly, progression of mitral valve prolapse to severe regurgitation occurs in twice as many women as men.38 Calcification of the mitral annulus in individuals younger than 40 years constitutes a minor criterion in the cardiovascular system.
Aortic valve dysfunction probably represents a late event in the continuum of annuloaortic ectasia secondary to root degeneration. The aortic valve, like the mitral valve, may also be susceptible to calcification at an early age.38 Dilated cardiomyopathy beyond explanation by the associated valvular incompetence is likewise seen in patients with Marfan’s syndrome. The mutant FBN1 protein in the cardiac ventricles has been implicated, but overall, rates of cardiomyopathy are low. In a study of 234 patients with Marfan’s syndrome but without significant aortic or atrioventricular valvular disease, 17 (7.3%) had left ventricular enlargement, but none had an ejection fraction less than 25%.39
Aortic Disease
Aortic aneurysm and dissection are the most life-threatening manifestations of Marfan’s syndrome. The threat depends on age, with rupture and dissection rates increasing as the aortic root dilates.40,41 Because root dilatation at the sinuses of Valsalva can begin in utero, lifelong transthoracic echocardiographic monitoring is needed. For patients in whom the aortic root and ascending aorta are poorly visualized as a result of anterior chest deformity, computed tomographic angiography or magnetic resonance angiography (to avoid radiation exposure) is a viable substitute. In contrast to degenerative aneurysm, the dilatation in Marfan’s syndrome may be confined to just the aortic root and not the ascending aorta. Normal aortic dimensions can vary widely with both age and size, and thus, their proper interpretation in patients affected by Marfan’s syndrome requires age-dependent nomograms.40 Absolute thresholds for replacement of the aortic root in children have not been established, given the observation that dissection is very rare in the young.5 However, if the aortic root is noted to grow more than 1 cm over consecutive annual assessments or if significant aortic regurgitation is present, early surgery may be necessary.31 In children and teenagers, a nomogram reflects the number of standard deviations of the patient’s aortic root from the mean aortic root diameter in the population and is termed a Z-score. If the child’s Z-score deviates rapidly from the population (>2 to 3 SD) under surveillance, aortic root repair may be justified to prevent rupture. In adults, surgical repair of the aortic root and ascending aorta to prevent aortic rupture and dissection is recommended when its greatest diameter exceeds 50 mm.31,41 Earlier intervention may be warranted with a family history of aortic dissection at lesser diameters.
Prevention
Lifestyle modifications are routinely recommended once the diagnosis of Marfan’s syndrome is established. On the basis of data from the United States, genetic cardiovascular diseases account for 40% of deaths in young athletes.42,43 A consensus document stated that “burst” exertions such as sprinting, weightlifting, basketball, and soccer should generally be avoided. Favored are recreational sports in which energy expenditure is stable and consistent over long periods, such as informal jogging, biking, and lap swimming.44 Symptoms potentially referable to a cardiovascular cause, such as shortness of breath, presyncope, and chest discomfort, should prompt immediate withdrawal from activity and evaluation. Litigation results suggest that physician reliance on consensus statements to determine medically reasonable levels of activity in patients with cardiovascular abnormalities is appropriate.45
Medical Treatment
Medical treatment with β-adrenergic receptor blockade to delay aortic root growth or prevent aortic dissection in patients with Marfan’s syndrome is currently considered a standard of care.5,31 General recommendations are a resting heart rate lower than 70 beats/min and a heart rate less than 100 beats/min with submaximal exercise. The rationale for this treatment paradigm is focused on decreasing proximal aortic shear stress and dP/dt. The only randomized trial assessing the effect of beta blockade studied 70 patients, 32 of whom received propranolol.46 Treated patients received titrated dosages via an open-label approach to keep the heart rate at 100 beats/min during exercise or a 30% rise in the systolic time interval. Serial echocardiograms were performed and correlated with age, height, and weight over a mean follow-up of 7 years. Fewer patients in the propranolol-treated group reached the primary endpoint of aortic regurgitation, aortic dissection, surgery, heart failure, or death (5 in treatment group and 9 in control group). Aortic growth after normalization was lower in the propranolol-treated group (0.023 cm/yr) than in the control group (0.084 cm/yr, P < .001).46 For patients with increased body weight or an end-diastolic aortic diameter greater than 40 mm, the response to beta blockade was worse, thus suggesting that beta blockers must be given at an adequate doses and early in the course of the disorder to optimize benefit.31,46 Approximately 10% to 20% of patients are intolerant of beta blockade because of asthma, fatigue, or depression. For these patients, a calcium channel blocker may be justified.47
The 2006 publication of the ability of losartan, a U.S. Food and Drug Administration–approved antihypertensive medication and selective angiotensin II receptor antagonist, to inhibit aortic aneurysm in murine models of Marfan’s syndrome48 has opened a potential new medical treatment paradigm. The exact mechanisms by which losartan inhibits TGF-β in the aortic wall are not completely understood, but a variety of feedback mechanisms are likely.49 A multicenter trial comparing beta blockade and losartan therapy for control of aortic root growth in children and young adults with Marfan’s syndrome has been initiated through the National Institutes of Health. Currently, there are no specific recommendations for losartan dosing or long-term therapy in individuals with Marfan’s syndrome.
Surgical Treatment
The traditional threshold for prophylactic surgical repair of the aortic root is 5 cm in patients with Marfan’s syndrome. The association between aortic diameter and risk for aortic catastrophe is clearly established, and aortic aneurysm size greater than 6 cm predicts a fourfold increase in risk for aortic rupture or dissection in patients with Marfan’s syndrome.50 For patients with acute manifestations—rupture in the aortic root or type A dissection—the goal of the surgery is preservation of life, and the most expedient repair may be placement of a graft with a mechanical valve (composite root replacement, Bentall procedure), as opposed to the more erudite repairs offered in elective scenarios to preserve the aortic valve by resuspension or remodeling techniques. Historically, aortic root catastrophe was the suspected culprit in 90% of Marfan-associated deaths,10 but once a dissection has occurred, degeneration to aneurysm may affect other segments of the aorta or its branches in survivors. Operations on the arterial tree outside the ascending aorta have been reported (Fig. 143-3) and can have acceptable outcomes.51 Indeed, as the life expectancy of individuals affected by Marfan’s syndrome has increased with prophylactic root replacement, it is plausible that the remaining aorta, or other large arteries, may progress to require repair in the absence of antecedent dissection. For aortic arch and descending thoracic or thoracoabdominal aneurysms, standard criteria for repair generally follow that of atherosclerotic aneurysms—a threshold of 5.5 to 6.0 cm.31
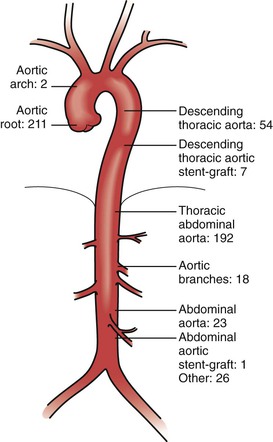
Figure 143-3 Distribution of vascular repairs in 300 patients with Marfan’s syndrome. (From Neglén P, et al: Stenting of the venous outflow in chronic venous disease: long-term stent-related outcome, clinical, and hemodynamic result. J Vasc Surg 46:979-990, 2007.)
Aortic Root
Composite surgical replacement of the aortic root and valve was pioneered by Bentall et al52 in 1968. Previously, the outlook for repair of dissection or aneurysms in the aortic root in patients with Marfan’s syndrome was dismal, with high bleeding rates and excessive mortality. The Bentall technique has proved to be safe and durable. In 1999, Gott et al41 reported the outcome of 675 patients with Marfan’s syndrome who underwent root and valve replacement between 1968 and 1996 at 10 centers with special expertise. Eighty-nine percent of patients received a composite valve graft (CVG), with 30% of the operations performed for acute type A dissection. Because of the risk of thromboembolism and the lifetime requirement for anticoagulation with warfarin, effort had been directed at maintaining the native aortic valve. This valve-sparing root replacement approach has the additional benefit of avoiding warfarin embryopathy in women with Marfan’s syndrome who may later desire pregnancy. To date, there has been no randomized trial comparing composite valve graft surgery and valve-sparing root replacement techniques, but the available data are encouraging and have shown low rates of valve dysfunction (20% to 25% with significant aortic regurgitation) and few reoperations.
Descending Thoracic and Thoracoabdominal Aorta
The first successful replacement of the thoracoabdominal aorta in a patient with Marfan’s syndrome was performed by Crawford53 in the 1980s. Elective surgical repair of descending thoracic aortic aneurysm and thoracoabdominal aortic aneurysms (TAAAs) in Marfan’s syndrome has benefited from the general refinements and introduction of adjuncts to reduce spinal cord injury and other major complications in a manner similar to standard atherosclerotic aneurysms (see Chapter 135). Prophylactic aortic replacement is indicated when the aortic diameter reaches 5.5 to 6.0 cm or if symptoms related to the aneurysm occur. Because of the frequent involvement of the descending and thoracoabdominal aorta with aneurysms of chronic dissection etiology, the extent of repairs in Marfan’s syndrome tend to be greater, with 42% to 78% of all TAAAs being DeBakey type II.51,54As expected, the mean age of patients with Marfan’s syndrome undergoing such repair is 34 to 48 years, younger than patients with non–connective tissue diseases.51,54,55 In comparison with patients undergoing repair of degenerative TAAAs, the rate of acute development of dissection or rupture in patients with Marfan’s syndrome and TAAAs was not higher (6% to 8% for each).51,55 Paraparesis and paraplegia rates after TAAA repair in Marfan’s syndrome compare favorably with those for non–connective tissue disease when matched for the extent of repair required.51,54,55 Because of the very young mean age of patients with Marfan’s syndrome undergoing TAAA repair versus the older mean age of patients with degenerative TAAA, overall long-term survival was better in those with Marfan’s syndrome.51
Given the preponderance of type II TAAA repairs in the available series, the rate of freedom from further aortic repair is high because little aorta remains to degenerate. However, secondary aortic procedures after TAAA repair in patients with Marfan’s syndrome are often performed for pseudoaneurysm or for aneurysm degeneration of the inclusion or Carrel patch (Fig. 143-4). Indeed, in the series reported by Lemaire et al,51 95% of reoperations (19 of 20) after previous TAAA repair (n = 178) in patients with Marfan’s syndrome were performed for visceral patch aneurysm.51 In our series of 107 patients who underwent TAAA repair, including creation of visceral patches, 17 were known to have a connective tissue disease.58 With a mean time to diagnosis of 6.5 years, 3 of these 17 patients (17.6%) were found to have aneurysmal degeneration of the visceral patch. By comparison, visceral patch aneurysms were noted after only 5.6% of atherosclerotic TAAA repairs.56All of these patients with Marfan’s syndrome had inclusion patches that encompassed the celiac axis, superior mesenteric artery, and both renal arteries, thus suggesting that the visceral patch should have been much smaller in all patients with connective tissue diseases to prevent late degeneration. I, along with many surgeons, avoid patch inclusion entirely and use a prefabricated four-branch graft to perform individual bypasses to the renal and visceral aortic branches. Intercostal inclusion patches that degenerate to aneurysms may be treated with stent-graft therapy, although paraplegia concerns are paramount. Given the morbidity associated with repair of the patch aneurysm (two intraoperative deaths in five of our patients taken to the operating room), Dardik et al56 recommend maintaining an indication for repair of 6.0 cm or larger.
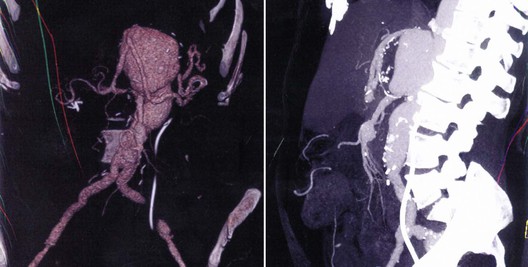
Figure 143-4 Ten-centimeter visceral patch aneurysm 7.5 years after a type II thoracoabdominal aortic aneurysm repair in a 48-year-old woman with Marfan’s syndrome. A hybrid open/endovascular stent-graft approach was used to repair the region. VRT reconstruction (left); lateral view demonstrating effacement of distance between SMA and celiac axis (right).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


