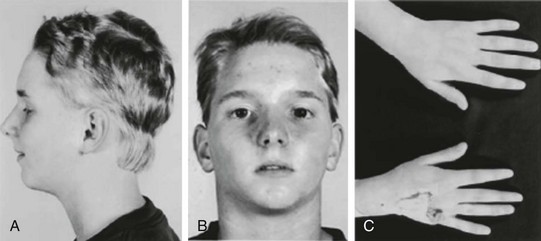
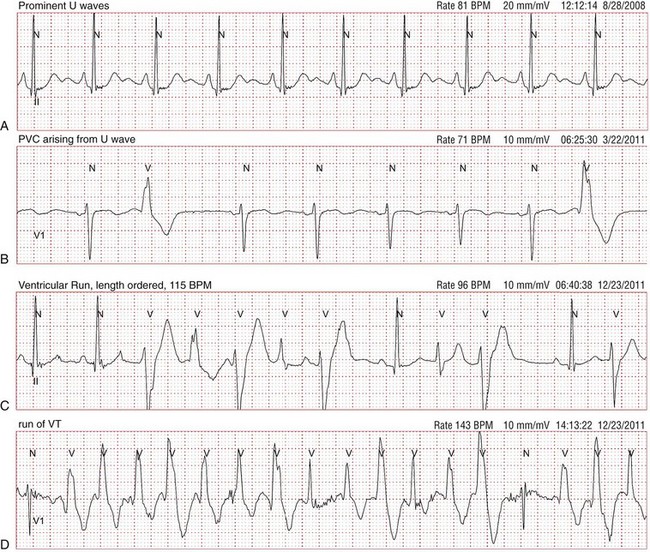
94 In 1971, Andersen et al.1 reported a series of patients with periodic skeletal muscle paralysis, ventricular ectopy, and dysmorphic features, the triad of clinical manifestations known as Andersen syndrome. Prolongation of the QT interval was incorporated as an important cardiac manifestation in subsequent larger studies of this disorder.2 The syndrome was renamed Andersen-Tawil syndrome (ATS) in recognition of the exceptional contributions of the clinical neurologist Rabi Tawil. Within the cardiovascular field, this disorder remained obscure until mutations in the K+ channel gene KCNJ2 were identified as a cause of ATS.3 KCNJ2 encodes the inward rectifier K+ channel Kir2.1, a component of the inward rectifier current IK1. IK1 provides substantial repolarizing current during the most terminal repolarization phase of the cardiac action potential, and it is the primary conductance controlling the diastolic membrane potential.4 The clinical features of ATS represent a spectrum of phenotypic manifestations encompassing the skeletal muscle and cardiac systems, in addition to craniofacial and skeletal anomalies (Figure 94-1). A major obstacle in the clinical diagnosis of ATS is the high degree of phenotypic variability and nonpenetrance. The full triad of clinical features (ventricular arrhythmias, periodic paralysis, and characteristic dysmorphic features) is present in 58% to 78% of mutation-positive patients,5 whereas between 32% and 81% manifest involvement of two of the three organ systems.3,5,6 Nonpenetrance ranges from 6% to 20% of mutation-positive individuals.5,6 In an interesting report, a large ATS family presented with sex-specific clinical manifestations. In this kindred, females manifested ventricular arrhythmias, whereas affected males had periodic paralysis. None of the 41 mutation carriers expressed the complete triad of ATS features.6 These sex-specific clinical findings, however, appear to be unique to this particular family in that the same mutation was reported in other individuals who manifested the typical ATS triad.7 Recently, the ATS phenotype was expanded to include several consistent craniofacial features and dental and skeletal anomalies that were observed in all members of a small cohort, regardless of cardiac or neuromuscular findings.8 These new findings help to clinch the clinical diagnosis when cardiac or skeletal muscle findings are absent. Figure 94-1 Typical dysmorphic features in a patient with Andersen-Tawil syndrome. Note the low-set ears, hypertelorism (wide interpupillary distance), micrognathia (small jaw) (A, B), and clinodactyly of the fifth digits (C). (Reprinted with permission from Plaster NM, Tawil R, Tristani-Firouzi M, et al: Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105:511–519, 2001.) ATS is an autosomal dominant or sporadic disorder of ventricular repolarization manifested by mild QTc interval prolongation, but marked prolongation of the QUc interval5,9 (Figure 94-2). Prominent U waves are commonly described. Zhang et al.9 reported distinct electrocardiogram (ECG) findings unique to ATS that included prolongation of the T wave downslope, wide T-U junction, high amplitude, and broad U waves.9 Arrhythmias in patients with ATS include frequent premature ventricular contractions, bigeminy, and polymorphic ventricular tachycardia (VT).5 Typically, VT is not sustained and bidirectional in nature with heart rates in the range of 130 to 150 beats/min. The degree of ventricular ectopy is highly variable between subjects, but up to 50% of all beats could be ventricular in origin.8 Although tachycardia burden is often high in patients with ATS,10 degeneration into lethal ventricular arrhythmias is relatively uncommon.11 For example, torsades de pointes was documented in only 3 of 96 KCNJ2 mutation-positive individuals.9 Figure 94-2 Representative electrocardiogram traces from patients with Andersen-Tawil syndrome (ATS). A, Rhythm strip demonstrating pathologic U waves at a heart rate of 81 beats/min. B, It is not unusual to observe premature ventricular contractions arising from the prominent U waves. C, A short run of nonsustained bidirectional ventricular tachycardia (VT; note alternating QRS axis polarity). Unlike catecholaminergic polymorphic ventricular tachycardia, bidirectional VT in ATS is typically slow (110 to 150 beats/min). D, A run of nonsustained polymorphic VT is demonstrated in this rhythm strip. In skeletal muscle, KCNJ2 mutations can cause intermittent muscle membrane inexcitability leading to episodic weakness that can last several hours.11 These episodes are clinically identical to those observed in other forms of periodic paralysis and are most frequently triggered by rest following exercise. Although other forms of periodic paralysis are clinically defined by serum potassium levels during an episode of paralysis, ictal serum potassium levels in ATS are commonly low, but can be normal or elevated in some patients.11 Episodes of periodic paralysis can be as infrequent as once per year to once per day. In addition to the episodic weakness, some patients with ATS can develop fixed, progressive weakness. A number of craniofacial and skeletal anomalies are described in ATS, the most common being low-set ears, ocular hypertelorism, broad nasal root, small mandible, clinodactyly in the hands, and syndactyly in the toes.2,5 Dental anomalies reported in ATS include persistent primary dentition, multiple missing teeth, and crowded dentition.8 Because of the phenotypic variability in ATS, the clinical diagnosis can be missed easily. Cardiologists should consider the diagnosis of ATS in patients with otherwise unexplained ventricular arrhythmias or long QT syndrome, and neurologists should screen all patients with periodic paralysis for cardiac involvement. In general, bidirectional VT is rarely observed in clinical medicine, but it is the hallmark of a handful of disease states, including ATS, catecholaminergic polymorphic VT (CPVT) and digitalis toxicity. Each of these clinical entities shares a common underlying abnormality in calcium homeostasis that can trigger delayed afterdepolarization-dependent arrhythmias. CPVT is characterized by sympathetic-mediated bidirectional or polymorphic VT and is due to mutations in calcium handling genes RYR2 (cardiac ryanodine receptor) and CASQ2 (calsequestrin-2).12 In a minority of KCNJ2 mutation-positive patients, bidirectional VT is enhanced by exercise, suggesting a clinical diagnosis of CPVT, especially in patients that lack other typical ATS features. Indeed, several patients have been reported with clinical manifestations that overlap ATS and CPVT, including patients submitted for commercial CPVT genetic testing.13–16 The KNCJ2 mutation V227F results in channels that function normally under basal conditions, but fail to increase current in response to protein kinase A stimulation, suggesting a clinical link between adrenergic stimulation and arrhythmia in the patient with ATS who is harboring this mutation.16
Andersen-Tawil Syndrome
Andersen-Tawil Syndrome
Clinical Manifestations of Andersen-Tawil Syndrome
Phenotypic Overlap Between Andersen-Tawil Syndrome and Catecholaminergic Polymorphic Ventricular Tachycardia
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thoracic Key
Fastest Thoracic Insight Engine
