Alveolar Hemorrhage Syndromes
Diffuse alveolar hemorrhage (DAH) is a potentially catastrophic complication of myriad immune and nonimmune disorders.1–3 Clinical features are broad, but hemoptysis, infiltrates on chest radiographs, hypoxemia, and progressive respiratory insufficiency are common to diverse etiologies.2 Nonimmune causes of alveolar hemorrhage include endobronchial tumors, arteriovenous malformations or aneurysms, ulcerative tracheobronchitis, hemorrhagic pneumonia, bronchiectasis, congestive heart failure, uremia, thrombocytopenia or coagulopathy, pulmonary venoocclusive disease, infections, and massive pulmonary embolism.4,5 These nonimmune causes need to be excluded in patients with severe alveolar hemorrhage. Depending upon the clinical scenario, coagulation profiles and ancillary tests (e.g., echocardiogram, chest computed tomographic [CT] pulmonary angiography, fiberoptic bronchoscopy) may be required to establish a specific diagnosis. In addition, other causes of diffuse parenchymal infiltrates (but without severe alveolar hemorrhage) share features in common with DAH syndromes (e.g., cryptogenic organizing pneumonia, hypersensitivity pneumonia, pulmonary alveolar proteinosis, and diverse interstitial or alveolar lung disorders). A discussion of these disorders is beyond the scope of this chapter, which focuses primarily on immune-mediated causes of DAH.
AUTOIMMUNE CAUSES OF ALVEOLAR HEMORRHAGE: DIFFERENTIAL DIAGNOSIS
Autoimmune DAH results from diffuse injury to the pulmonary microvasculature (termed capillaritis or endotheliitis) (Table 68-1).6,7 Systemic necrotizing vasculitides8,9 (principally microscopic polyangiitis [MPA]10,11 and Wegener’s granulomatosis 12 account for most cases of autoimmune DAH.* Other causes of autoimmune DAH include antiglomerular basement membrane (anti-GBM) antibody disease,13–15 connective tissue disease (CTD) (principally systemic lupus erythematosus [SLE]),16 exogenous agents or drugs.17 In many of these disorders, rapidly progressive glomerulonephritis (RPGN) is present concomitantly.18,19 In most patients with autoimmune DAH and glomerulonephritis (GN), anti-GBM antibody and immune complexes are lacking.19 The term pauci-immune glomerulonephritis has been used to refer to this group of patients, who encompass a heterogeneous group of disorders.20,21 (discussed in detail below).
Idiopathic pulmonary hemosiderosis (IPH), a rare cause of recurrent DAH with no renal or extrapulmonary component, occurs primarily in children22–24 and remains a diagnosis of exclusion.
Differentiation of these diverse syndromes can usually be accomplished by serological studies and by kidney biopsy. In such cases, lung biopsy is not required. GN can be demonstrated in the great majority of patients with DAH complicating granulomatosis with polyangiitis (GPA)12 or MPA.7,10,11 By contrast, the kidneys may be spared in DAH associated with CTD,16 bone marrow transplant recipients,25,26 or immunocompromised patients.27,28 Urinalysis (to look for microscopic hematuria, red cell casts, and proteinuria) and measurement of renal function should always be done in the diagnostic evaluation of DAH. Findings consistent with GN warrant a prompt and aggressive evaluation that should include percutaneous needle biopsy of the kidney.
CLINICAL FEATURES OF AUTOIMMUNE ALVEOLAR HEMORRHAGE
Irrespective of etiology, the clinical, radiographic, and histopathological features of DAH may be similar.1,2,6 Classical findings are hemoptysis, diffuse alveolar infiltrates, hypoxemia, renal failure, and iron-deficiency anemia.2,7 However, the clinical spectrum is wide, and many of these features may be subtle or absent. In this context, the diagnosis of DAH may be difficult, as signs and symptoms overlap with diverse etiologies of diffuse alveolar infiltrates. Prompt diagnosis and institution of therapy is vital to avert early mortality from DAH and late sequelae from end-stage renal failure. Chest radiographs typically reveal bilateral alveolar infiltrates, often with a batwing appearance. However, focal, and even unilateral, patterns indistinguishable from pneumonia may occur. Following cessation of bleeding, infiltrates markedly improve or normalize within 24 to 72 hours (Fig. 68-1). A presumptive diagnosis of DAH can often be made by a combination of clinical and serological findings and bronchoalveolar lavage (BAL) fluid. Grossly bloody BAL fluid (with progressively more blood with serial aliquots),1,2,7 large numbers of hemosiderin-laden macrophages,8,27 and the absence of purulent secretions or ancillary evidence for infection strongly support DAH as a cause of pulmonary infiltrates. Ancillary studies including serologies, renal function tests, and urinalysis may support the diagnosis.
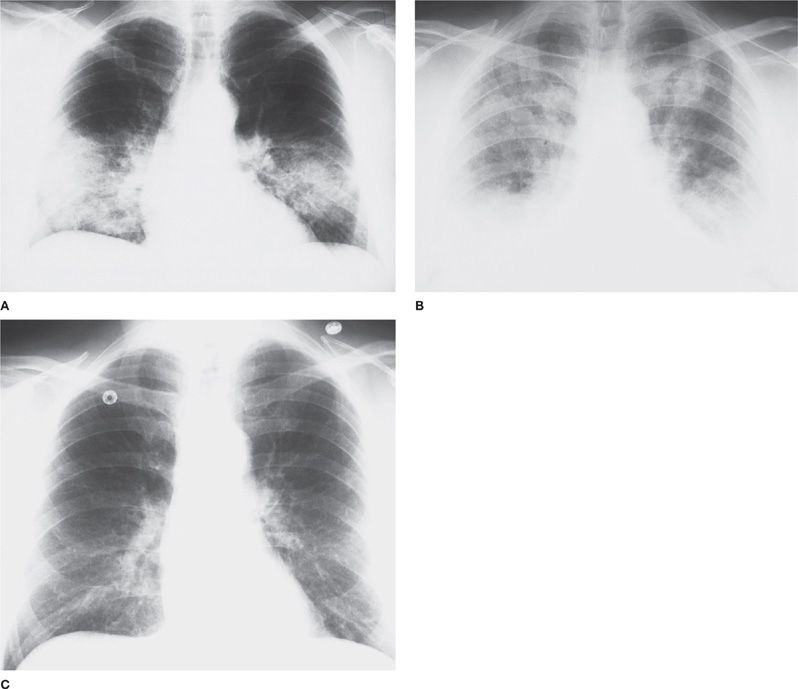
Figure 68-1 A. Idiopathic rapidly progressive glomerulonephritis. Posterior–anterior (PA) chest radiograph from a 52-year-old man with rapidly progressive glomerulonephritis, hemoptysis, and bilateral alveolar infiltrates, consistent with alveolar hemorrhage. Bronchoalveolar lavage demonstrated blood-tinged fluid and numerous hemosiderin-laden macrophages. B. Idiopathic rapidly progressive glomerulonephritis. PA chest radiograph from the same patient 18 months later with diffuse bilateral alveolar infiltrates representing recurrent massive alveolar hemorrhage. He was treated with pulse methylprednisolone (1 g daily for 3 days), followed by a gradual corticosteroid taper. C. PA chest radiograph from the same patient 3 weeks later demonstrating complete resolution of the alveolar infiltrates.
DIAGNOSIS
 THE ROLE OF LUNG BIOPSY
THE ROLE OF LUNG BIOPSY
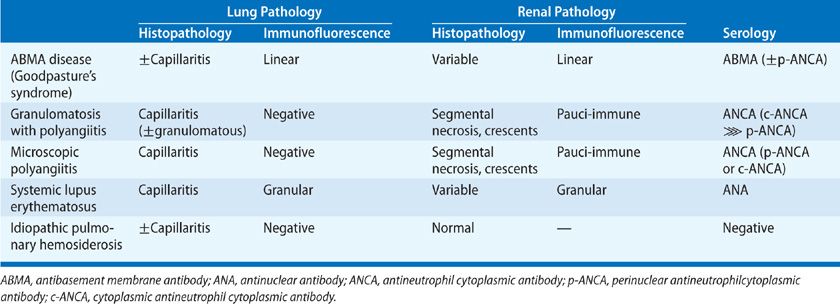
The role of lung biopsy in the diagnosis of DAH and the determination of its etiology is controversial. We believe the risks of open or thoracoscopic lung biopsy are excessive in patients with severe DAH and respiratory failure. Postoperative complications such as infection and air leaks may be exacerbated by the corticosteroid or immunosuppressive agents used to treat many of these immune-mediated DAH syndromes. Furthermore, histological features are usually nonspecific. Predominant findings are extensive intra-alveolar hemorrhage and necrotizing pulmonary capillaritis (endotheliitis) (Fig. 68-2).6–8 Capillaritis is characterized by neutrophilic infiltration of capillaries, fragmented neutrophils (leukocytoclasis), and necrosis of the capillary walls (Fig. 68-3).8 Loss of the integrity of the alveolar–capillary basement membrane results in leakage of red blood cells and neutrophils into the alveolar space.2 Hemosiderin-laden macrophages (siderophages) accumulate within the alveolar spaces and interstitium; their presence is a clue to prior episodes of alveolar hemorrhage (Figs. 68-2B,C and 68-4).8
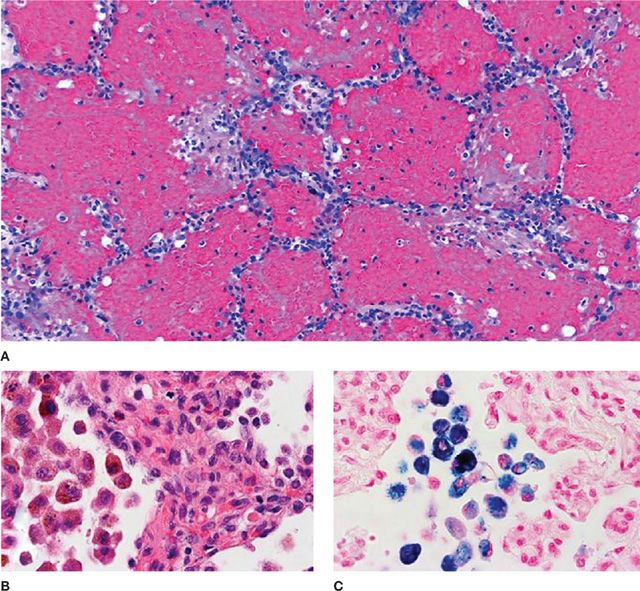
Figure 68-2 Alveolar hemorrhage. A. Acute hemorrhage with blood-filling alveolar spaces (H&E stain, ×40). B. Pigment-laden alveolar macrophages (H&E, ×200) shown in (C) to be full of iron (blue cytoplasm) indicative of prior hemorrhage (Prussian blue stain, ×200).
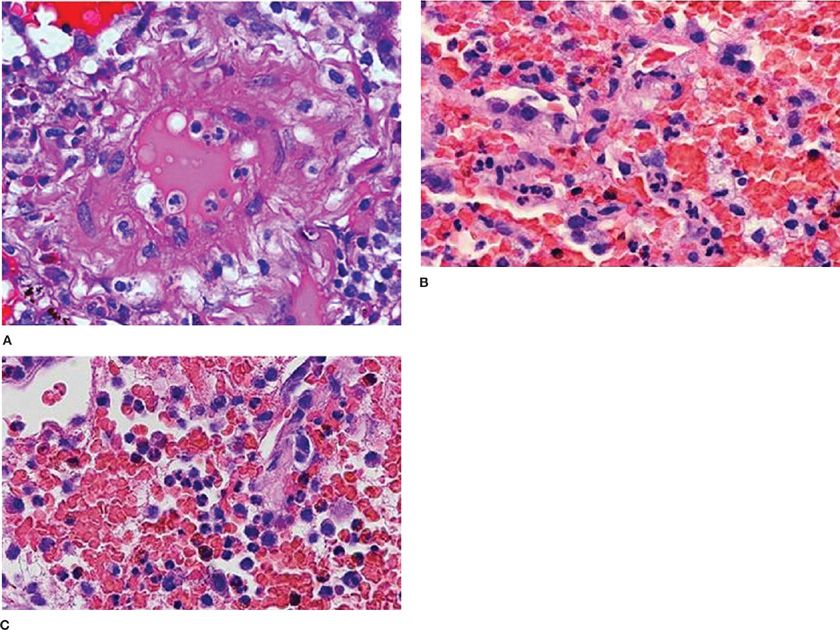
Figure 68-3 Pulmonary capillaritis. A. Necrotizing arteritis in microscopic polyangiitis (H&E, ×400); (B) capillaritis with intact alveolar septae in Wegener’s granulomatosis (granulomatosis with polyangiits) (H&E, ×400); and (C) capillaritis with destruction of alveolar septae (H&A, ×400).
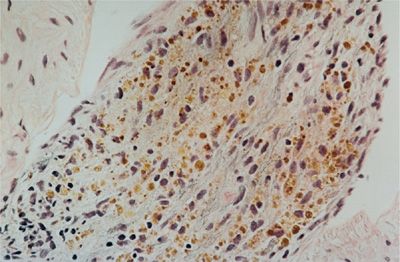
Figure 68-4 Hemosiderin-laden macrophages (siderophages) are prominent in the alveolar interstitium in a patient with recurrent alveolar hemorrhage (H&E). (Used with permission of Joseph Fantone, MD.)
Capillaritis was initially described as a marker of systemic vasculitis, but may also be observed in myriad disorders associated with DAH (e.g., SLE,16 CTD,2 anti-GBM disease,14 bone marrow25,26 or lung29 transplant recipients, and drug-induced DAH).1,6,7 An associated venulitis and arteriolitis may sometimes be present, but larger vessels are spared.8 Capillaritis is subtle and often overshadowed by DAH filling the alveolar spaces.
Pulmonary capillaritis can be diagnosed by transbronchial biopsy, but this diagnosis is made with greater confidence when larger biopsy specimens are obtained by video-assisted thoracoscopy or limited thoracotomy.6 Additional pathological features may be seen in patients with underlying granulomatous vasculitis (e.g., granulomas, necrosis, or eosinophils).6,8 Nongranulomatous inflammation in airways and lung interstitium, interstitial fibrosis, diffuse alveolar damage (DAD), fibrinous pleuritis, and organizing pneumonia have also been described in DAH associated with antineutrophil cytoplasmic antibody (ANCA) vasculitic syndromes.8 It should be emphasized that histological findings of alveolar hemorrhage and capillaritis, although distinctive, are nonspecific.7 Immunofluorescent (IF) stains (of lung or kidney) or serological markers (e.g., anti-GBM antibody or ANCA) are required to differentiate the various causes of autoimmune DAH (Table 68-2).6–8 Linear deposits of immunoglobulin G (IgG) along alveolar septa is pathognomonic for anti-GBM disease.13,14 A granular, or “lumpy-bumpy” pattern of immune complex deposits may be seen in SLE, systemic necrotizing vasculitis, or immune complex–mediated idiopathic RPGN.6 In ANCA-associated capillaritis, immune complexes are usually lacking (hence the term pauci-immune).10,11,30 When immune DAH is suspected, a portion of the lung biopsy can be frozen for IF stains, but IF stains of lung tissue are logistically difficult, and nonspecific background staining may lead to misinterpretation. When GN is present concomitantly, kidney IF stains are more sensitive and reliable.
Despite the greater accuracy of surgical lung biopsy in evaluating DAH, fiberoptic bronchoscopy with BAL is usually adequate to exclude infectious etiologies and support the diagnosis of DAH.1 Bloody or serosanguinous BAL fluid (consistent with active or recent bleeding)1,2,7 or hemosiderin-laden macrophages8,27 (a clue to prior episodes of alveolar hemorrhage) may be sufficient to justify initiation of therapy provided clinical and serological features are consistent. Thoracoscopic lung biopsy may be useful in noncritically ill patients with suspected DAH when ancillary studies, kidney biopsy, and BAL are nondiagnostic.
 THE ROLE OF PERCUTANEOUS KIDNEY BIOPSY
THE ROLE OF PERCUTANEOUS KIDNEY BIOPSY
Necrotizing GN is a cardinal (albeit nonspecific) feature of most immune-mediated DAH syndromes.19,31 The histological spectrum is varied, ranging from mild mesangial thickening to severe crescentic GN. Vasculitis of renal arterioles is rarely found, even in granulomatous vasculitides. Because of the strong association of autoimmune DAH and GN, percutaneous kidney biopsy should be performed in any patient with suspected DAH who has abnormalities on urinalysis or renal function tests. Conventional hematoxylin and eosin (H&E) stains are nonspecific, but the demonstration of glomerular inflammation with necrosis and crescents supports the diagnosis of an immune-mediated etiology (Fig. 68-5).19,31,32 IF stains may clarify the nature of the underlying disorder. Bright linear IF staining along glomerular basement membranes (GBMs) is pathognomonic for anti-GBM disease (Fig. 68-6).13,33 A lumpy-bumpy IF pattern, consistent with deposits of immune complexes, is found in some cases of CTD16 and in idiopathic immune complex–mediated GN.34 Negative IF stains are characteristic of the pauci-immune GN of necrotizing vasculitis.19,30,35,36 Serologies are critically important in defining the underlying disorder responsible for DAH (particularly ANCA, anti-GBM antibody, and antinuclear antibodies). Recognizing the different pathogenetic mechanisms of these DAH syndromes is important, as the prognosis and treatment strategies differ.
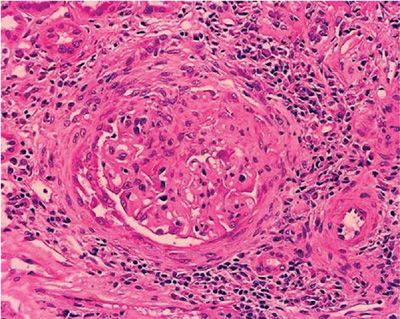
Figure 68-5 Segmental necrotizing and crescentic glomerulonephritis due to vasculitis (H&E).
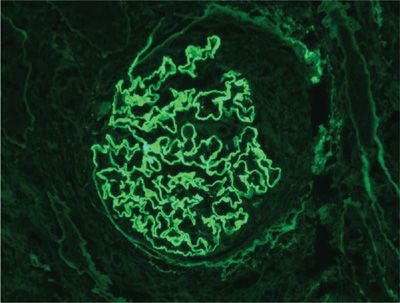
Figure 68-6 Linear immunofluorescent staining along glomeruli due to deposition of anti-glomerular basement membrane (anti-GBM) antibody.
 THERAPY OF IMMUNE-MEDIATED ALVEOLAR HEMORRHAGE
THERAPY OF IMMUNE-MEDIATED ALVEOLAR HEMORRHAGE
Irrespective of etiology, the most immediate concern in patients with severe immune DAH is to control intrapulmonary bleeding, which may be fatal. Besides general supportive measures, corticosteroids are considered part of standard therapy for all immune-mediated DAH syndromes. For severe cases (e.g., severe hypoxemia, respiratory failure), high-dose “pulse” methylprednisolone (1000 mg daily for 3 days) should be given (irrespective of underlying etiology), even while pursuing a diagnostic workup.1,7 Delaying pulse therapy in a critically ill patient for even a few hours may be catastrophic. Rapid resolution of bleeding can occur, often within 24 to 72 hours of initiation of therapy (Fig. 68-7). Following the 3-day pulse, corticosteroids (dose of methylprednisolone 60 to 120 mg per day or equivalent) should be continued for a few days, until control of the bleeding and extrapulmonary manifestations has been achieved. The subsequent dose and rate of corticosteroid taper need to be individualized, based upon clinical, radiographic, and serological response. The presence of renal involvement, vasculitis, or progression of DAH on corticosteroids is an indication for adding cyclophosphamide (CYC) (or occasionally other immunosuppressive agents).1,18,37 Rituximab may be as effective, and possibly more effective than CYC for ANCA-associated vasculitis,38,39 but data are limited for DAH. Plasmapheresis is a central component of therapy for anti-GBM disease13,14 but has no routine role for other disorders.11,12,40,41 However, plasmapheresis may have an adjunctive role in patients with autoimmune DAH and severe renal insufficiency (i.e., serum creatinine >4 mg%)41,42 and in patients with severe or progressive DAH refractory to corticosteroids or immunosuppressive agents.43 Measures to ensure adequate oxygenation are also essential. Mechanical ventilatory support, often with positive end-expiratory pressure, may be necessary in fulminant cases of DAH, to prevent death due to refractory hypoxemia. Transfusion of red blood cells may be required to maintain an acceptable hematocrit (more than 25%) and adequate blood pressure. In the sections that follow, we discuss each of the autoimmune DAH syndromes individually.
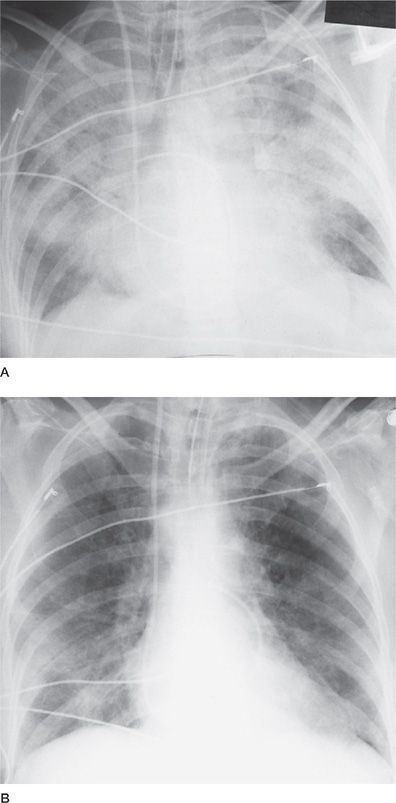
Figure 68-7 A. Alveolar hemorrhage due to microscopic polyarteritis (MPA). Posterior–anterior (PA) chest radiograph demonstrating massive alveolar infiltrates involving all lobes. Because of the severity of respiratory failure (requiring 16 cmH2O of positive end-expiratory pressure to achieve acceptable oxygenation), no lung biopsy was performed. Urinalysis demonstrated numerous red cells and occasional red cell casts. Serum creatinine was 1.4 mg%. Pulse methylprednisolone (1 g daily × 3 days) was initiated, and renal biopsy was scheduled for the following morning. B. Alveolar hemorrhage due to MPA. PA chest radiograph from the same patient 12 hours following initiation of pulse methylprednisolone. Marked improvement in alveolar infiltrates is evident. Renal biopsy demonstrated glomerulonephritis and a necrotizing vasculitis involving renal arterioles; no granulomas were present. Cyclophosphamide (2 mg/kg per day) was instituted, and corticosteroids were continued. Within 5 days, the infiltrates had cleared completely and serum creatinine was 0.6 mg%.
SPECIFIC SYNDROMES
 GOODPASTURE’S SYNDROME
GOODPASTURE’S SYNDROME
Clinical Features
Anti-GBM disease (Goodpasture’s syndrome), the prototype of pulmonary renal syndromes, accounts for 18% to 32% of immune-mediated DAH.3,13,15,31,44 Classically, anti-GBM disease manifests as DAH and RPGN.13,14,33,45 In 30% to 50% of patients with anti-GBM disease, GN occurs without DAH; DAH alone is rare (<5%).14,15 Anti-GBM disease typically affects individuals between 20 and 45 years of age with a distinct male predominance.14,45 The incidence has been estimated as 1 to 3 cases per million population per year.46,47 The etiology is not known, but exposure to inhaled hydrocarbons,46 cigarette smoke,45 cocaine,15 and antecedent viral illnesses, particularly influenza, have been cited as risk factors.14 The demonstration of anti-GBM antibodies in tissue (typically kidney)33 or in serum13,14 is the cornerstone of the diagnosis.
The clinical expression of anti-GBM disease is highly variable. Most patients present with progressive dyspnea, diffuse alveolar infiltrates, and hypoxemia; hemoptysis occurs in 45% to 94%.13–15,48 GN is a cardinal feature.33,47,49,50 Microscopic hematuria, red cell casts, or proteinuria are almost always present. Gross hematuria occurs in up to 41% of patients.14 Azotemia is noted in 55% to 71% of patients at presentation.13,48 Fatigue and weakness are common.14 In the absence of therapy, progressive renal insufficiency ensues, often resulting in end-stage renal failure within days to weeks of the onset of symptoms.13 Oliguria, severe renal failure, or greater than 50% crescents on renal biopsy are associated with a poor prognosis and low rate of recovery of renal function.13,14,33,51 The course may be fulminant, with severe renal failure and explosive, life-threatening DAH.13,14 Chest radiographs typically reveal dense bilateral alveolar infiltrates, often with air-bronchograms. With cessation of bleeding, infiltrates may resolve within 24 to 36 hours. Pleural effusions are rare and suggest an alternative diagnosis. Pulmonary function tests are rarely helpful in the acute setting of DAH.14,15 Bloody or serosanguineous BAL fluid (that worsens with serial aliquots) suggests DAH but is nonspecific.8 Anemia is present in more than 90% of cases and may be profound. Serum iron and ferritin levels are usually decreased, reflecting diminished iron stores. Factors associated with a higher incidence of DAH include cigarette smoking,15 exposure to high concentrations of oxygen, upper respiratory tract infections, and increased hydrostatic (pulmonary capillary) pressures.13,14
Serological assays for anti-GBM antibody are invaluable in confirming the diagnosis and monitoring the adequacy of therapy.14,47 Radioimmunoassays or enzyme-linked immunosorbent assays (ELISA) for anti-GBM antibody are highly sensitive (>95%) and specific (>97%)13,14 but are performed in only a few laboratories. Results are usually not available for several days. Since delay in institution of therapy may preclude a favorable outcome, percutaneous renal biopsy is usually performed while awaiting the results of serum assays. Although disparate results have been reported, the height of serum anti-GBM antibody titer correlated with the prognosis of renal disease in some studies.48,52 Further, changes in anti-GBM titer over time may be a guide to efficacy of therapy.14 Treatment can be tapered and discontinued after the antibody has disappeared from the circulation (usually within 3–6 months).15 Up to one-third of patients with circulating anti-GBM antibodies have concomitant ANCA (usually MPO).13,32,53 Other serological studies are negative or nondiagnostic.
 HISTOPATHOLOGY
HISTOPATHOLOGY
Percutaneous kidney biopsy is the preferred invasive procedure to substantiate the diagnosis of anti-GBM disease.15 Light microscopy demonstrates a proliferative or necrotizing GN, often with cellular crescents.14,33 Over time, the crescents may fibrose, and frank glomerulosclerosis, interstitial fibrosis, and tubular atrophy may be observed.33 Although these microscopic features are nonspecific, IF stains are the cornerstone of the diagnosis. Bright linear deposits of IgG and complement (C3) along GBM are pathognomonic of anti-GBM disease (Fig. 68-6).14,33 All four subclasses of IgG are represented, but IgG154,55 and IgG355 predominate in anti-GBM disease. Rare cases of linear deposits of IgM or IgA have been described.15 Lung biopsies are rarely necessary,14 as the histological features on renal biopsy are usually adequate to establish the diagnosis. When lung biopsy has been done, extensive hemorrhage predominates6,8 with accumulation of hemosiderin-laden macrophages within the alveolar spaces.14 Foci of DAD and capillaritis may also be found.6 Interstitial or intra-alveolar inflammation is minimal or absent. Extensive necrosis or large-vessel vasculitis is not found. Similar histopathological features may be seen with a wide gamut of immune-mediated DAH syndromes.6,8 IF stains of lung tissue may be diagnostic, provided a clear linear pattern of immunofluorescence is present. However, IF stains are technically difficult in lung tissue and autofluorescence may obscure the linear IgG deposits.
 PATHOGENESIS
PATHOGENESIS
Antibodies are directed against the α3 chain of type IV collagen, an antigen highly expressed in both alveolar and GBMs.13,15,54 Anti-GBM antibodies bind the GBM and activate complement, initiating an inflammatory pathway that elicits injury.13 In addition to circulating antibodies, autoreactive T lymphocytes directed against the α3 antigen are key mediators for development of RPGN.15,56 Immunoglobulin synthesis and deposits of IgG along the alveolar and glomerular capillary basement membranes then ensue. Anti-GBM disease is monophasic, and during the course of the disease, self-tolerance is restored.13 Late relapses are rare.13 This tolerance may be achieved by regulatory CD4+ and CD25+ T cells15,57 or anti-idiotypic (blocking) antibodies,54 but this remains speculative.
The pathogenesis of anti-GBM disease is unknown, but both genetic15,58 and environmental factors54 may play roles. Patients with anti-GBM disease preferentially express certain immunoglobulin Gm allotypes and links between anti-GBM disease and the HLA-DR2 histocompatibility antigen have been noted.13,15 Exposure to cigarette smoke, hydrocarbon-containing solvents, hard-metal dust, influenza A2 virus, chlorine gas, and D-penicillamine have been associated with anti-GBM disease.13–15 These exogenous factors may injure the basement membrane, resulting in increased capillary permeability, exposing the Goodpasture’s antigen (α3 chain), which is then recognized as foreign, eliciting a T-helper cell response.15,54
 TREATMENT
TREATMENT
Before the availability of the current therapy and renal dialysis, mortality exceeded 90%.13 Plasmapheresis was introduced as a therapeutic option for anti-GBM disease in the 1970s,49 and was quickly adopted worldwide and incorporated in all clinical trials. Currently, with the combination of plasmapheresis, corticosteroids (CSs), and CYC, mortality has been reduced to less than 20%.13–15 Because of the rarity of anti-GBM syndrome, only one small randomized trial compared immunosuppressive therapy with the combination of immunosuppressive therapy plus plasma exchange.49 In that study, plasmapheresis plus immunosuppressive therapy was associated with more rapid disappearance of anti-GBM antibody and improved renal function than treatment with immunosuppressive agents alone.49 The optimal extent and duration of plasma exchanges have not been defined. Most investigators advocate plasma exchange daily or every other day for 2 to 3 weeks, until the clinical course has improved and serum anti-GBM antibodies are nondetectable.13,41 Immunosuppressive therapy is required to inhibit antibody production and rebound hypersynthesis, which may occur following discontinuation of plasma exchange.13 Treatment of acute, life-threatening DAH in Goodpasture’s syndrome is similar to other autoimmune disorders. Pulse methylprednisolone (1 g daily for 3 days) is given, followed by a gradual corticosteroid taper.14 Either CYC (oral or IV pulse) or azathioprine should be initiated once the diagnosis of anti-GBM disease is substantiated. Most investigators favor CYC over azathioprine,13–15 but studies comparing these agents have not been done. CYC is maintained for the duration of therapy, unless complications such as leukopenia necessitate dose reduction. The corticosteroid dose is gradually tapered over several months. Immunosuppressive or cytotoxic therapy may be discontinued within 3 to 6 months provided a sustained remission has been achieved and anti-GBM antibodies have disappeared.13,15 Circulating anti-GBM antibodies usually clear within 8 weeks, irrespective of the initial titer. Early relapse (within the first 2 months) may occur when circulating antibodies are still present. This typically manifests as DAH. Risk factors for relapse include infection, volume overload, and cigarette smoking.15 Late recurrence, associated with renewed antibody synthesis following a remission, is rare.14 In summary, aggressive therapy with plasmapheresis, corticosteroids, and immunosuppressive agents has dramatically improved prognosis.14,15,51 With this approach, 5-year survival exceeds 80%, and fewer than 30% of patients require chronic dialysis.14,15 Early recognition and treatment of this syndrome are critical, as the prognosis for recovery of renal function depends upon the initial extent of injury.32,50 Recovery of renal function can be expected in patients with minor functional impairment. By contrast, patients manifesting initial serum creatinine greater than 4 mg/dL, oliguria, or greater than 50% crescents on renal biopsy rarely recover and usually progress to end-stage renal failure requiring chronic dialysis.32,50 In one study of 71 patients with anti-GBM disease (all were treated with steroids, immunosuppressive agents, and plasmapheresis), renal survival was linked to extent of renal failure at presentation.51 Renal survival rates at 1 year were as follows: 8% among 39 patients requiring dialysis at presentation; 82% among 13 patients with creatinine >5.7 but not requiring dialysis; 95% among 19 patients with initial serum creatinine <5.7 mg%. Renal transplantation has been successful in patients with irreversible renal failure, provided serum anti-GBM antibodies are undetectable.59,60
Systemic Vasculitis
DAH is a common complication of MPA10,11 and granulomatosis with polyangiitis (GPA) (formerly called Wegener’s syndrome)12,61 and rarely complicates Churg–Strauss syndrome (CSS),62,63 Behçet disease,64 mixed cryoglobulinemia,65 Henoch–Schöenlein purpura,66 and other systemic necrotizing vasculitides.1,37 Classic polyarteritis nodosa (PAN) rarely involves the lung.8,67 Necrotizing small-vessel vasculitis accounts for the majority of autoimmune DAH syndromes.1,8 RPGN is usually present in each of these DAH syndromes, but the disease is sometimes limited to the kidneys or lungs. Circulating antibodies directed against cytoplasmic components of neutrophils and monocytes (ANCA) have been detected in most patients with these “pulmonary–renal syndromes,”7,34,37,68 suggesting a common pathogenesis and mechanism of lung injury in these diverse vasculitic disorders.
ANCA-Associated Vasculitides
Goodpasture’s syndrome (anti-GBM disease) was the first of the pulmonary–renal syndromes to be immunologically characterized.14,15 Subsequent studies documented immune complexes in serum or renal tissue in subsets of patients with pulmonary–renal syndromes, particularly SLE16 and immune complex–mediated GN.69 However, more than two-thirds of patients with pulmonary–renal syndromes are mediated by ANCA antibodies without anti-GBM or immune complexes (i.e., pauci-immune).1,34,37 Depending upon clinicopathological features, some patients with pauci-immune GN and DAH may meet criteria for GPA or CSS, while others exhibit a multisystemic small-vessel vasculitis but lack granulomatous inflammation of the respiratory tract. In this context, the term microscopic polyangiitis is used.10,11,70 The availability of serum assays for ANCAs has profoundly influenced the classification of immune DAH and GN. ANCA-positive patients with pauci-immune DAH and GN (formerly given a diagnosis of idiopathic RPGN and DAH) are now considered to have MPA.11 The spectrum of ANCA-associated diseases is not limited to patients with pulmonary–renal syndromes but includes individuals with MPA limited to the lung (i.e., manifesting as DAH) or kidney (i.e., necrotizing GN).11 To avoid further confusion, brief definitions of the major ANCA-associated vasculitides are outlined below.
 GRANULOMATAOSIS WITH POLYANGIITIS, GPA (FORMERLY CALLED WEGENER’S GRANULOMATOSIS)
GRANULOMATAOSIS WITH POLYANGIITIS, GPA (FORMERLY CALLED WEGENER’S GRANULOMATOSIS)
GPA, the most common of the pulmonary vasculitides, typically involves the upper respiratory tract (e.g., sinuses, ears, nasopharynx, oropharynx, trachea), lower respiratory tract (bronchi and lung), and kidney, with varying degrees of disseminated vasculitis (see Chapter 83).12 The annual incidence of GPA has been estimated at 4 to 12 cases per million.12,46,71,72 Alveolar hemorrhage is a rare complication of GPA, reflecting diffuse injury to the lung microvasculature (i.e., capillaritis) (Fig. 68-8).1,6 In this context, RPGN is present in more than 90% of patients.6,12 The salient histopathological features of GPA include small-vessel vasculitis (involving capillaries, arterioles, venules), geographic necrosis, hemorrhagic infarcts, a mixed inflammatory cellular infiltrate, and a granulomatous component.6,8,12 Circulating c-ANCAs (PR3 epitope) have been detected in more than 90% of patients with active generalized GPA and in 40% to 70% with active regional GPA.12,68,73–75 Oral CYC (2 mg/kg per day) plus prednisone has been the initial treatment of choice for GPA for more than 3 decades.12,76,77 With this regimen, remissions are achieved in 70% to 93% of patients, with early mortality rates of less than 15%.76–81 By 3 to 6 months, assuming complete remissions are achieved, azathioprine78,79 or methotrexate79 can be substituted for CYC. Treatment should be continued for a minimum of 12 to 18 months (total duration).12,77 Relapses can be treated with CYC and prednisone. Methotrexate may be used in patients with limited disease or those experiencing significant toxicity from CYC.82,83 Rituximab may be as effective, and possibly more effective than CYC for GPA and AAV.38,39,84–86 However, whether rituximab should be used with corticosteroids alone or corticosteroids combined with CYC has not been clarified. Further, the role for long-term maintenance therapy with rituximab has not been studied.84–86 Additional studies are required to assess indications for rituximab, appropriate dosing and frequency of administration, role for concomitant therapy, and long-term side effects. Trimethoprim/sulfamethoxazole may have an adjunctive role (together with CYC and prednisone) to reduce relapse rates,87 but should not be considered as primary therapy.
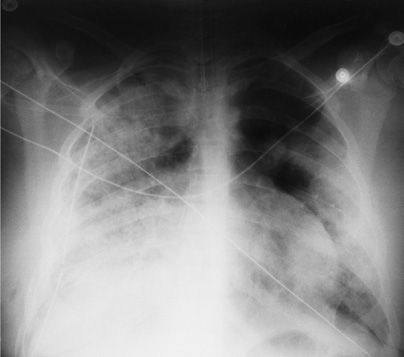
Figure 68-8 Granulomatosis with polyangiitis (GPA) (formerly called Wegener’s granulomatosis. Posterior–anterior (PA) chest radiograph demonstrated bilateral alveolar infiltrates in a 13-year-old girl with hemoptysis and respiratory failure. A right chest tube is in place from an open lung biopsy performed 2 days earlier. Open lung biopsy demonstrated capillaritis and massive alveolar hemorrhage. Pulse methylprednisolone, followed by oral cyclophosphamide and prednisone, was associated with a complete remission.
 CHURG–STRAUSS SYNDROME (ALLERGIC ANGIITIS AND GRANULOMATOSIS)
CHURG–STRAUSS SYNDROME (ALLERGIC ANGIITIS AND GRANULOMATOSIS)
CSS, also termed allergic angiitis and granulomatosis, is a rare, small-vessel vasculitis associated with a prominent allergic component, asthma, and eosinophils in blood or involved tissues (see Chapter 83).63,88 CSS involves capillaries, venules, and arterioles.63 Granulomas, eosinophils, and palisading histiocytes in extravascular tissues are hallmarks of the disorder88,89 and distinguish CSS from other vasculitides.8,63 The annual incidence of CSS has been estimated at 0.6 to 6.8 cases per million.46,71,88,90 In the classic form of CSS, vasculitis develops after a several-year history of atopy or asthma.89 Pulmonary involvement is nearly invariably present.63,88,89 Asthma is present in 96% to 100%; focal infiltrates on chest radiographs, in 30% to 70% of cases.63,88,89 DAH is a rare complication of CSS. In a French series of 112 patients with CSS, moderate DAH was observed in 3/43 ANCA-(+) patients and in 5/69 ANCA-negative patients.91 DAH was cited in only 1/32 patients in a Spanish series,92 and 1/19 in an Italian series.93 Severe DAH has only rarely been reported in CSS.94,95 Constitutional symptoms may herald the onset of vasculitis.88 Extrapulmonary manifestations include mononeuritis multiplex (63%–93%); skin (50%–78%); cardiac (16%–56%); kidney (16%–49%); skin (50%–78%); gastrointestinal (GI) tract (17%–58%).63,88 Factors associated with a worse prognosis include cardiac or GI tract involvement, renal insufficiency; age >65 years.96
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


