Aging and Cardiovascular Disease in the Elderly: Introduction
The world population in both industrialized and developing countries is aging. In the United States, 35 million people are older than the age of 65 years, and the number of older Americans is expected to double by the year 2030. The clinical and economic implications of this demographic shift are staggering because age is the most powerful risk factor for cardiovascular diseases.
The incidence and prevalence of hypertension, coronary artery disease, congestive heart failure, and stroke, the quintessential diseases of Western society, increase steeply with advancing age (Fig. 102–1). Although epidemiologic studies have discovered that some aspects of lifestyle and genetics are risk factors for these diseases, age, per se, confers the major risk. There is a continuum of age-related alterations of cardiovascular structure and function in healthy humans.1-4 These changes appear to influence the steep increases in hypertension, atherosclerosis, stroke, left ventricular hypertrophy, chronic heart failure, and atrial fibrillation with increasing age. Specific pathophysiologic mechanisms that underlie these diseases become superimposed on cardiac and vascular substrates that have been modified by an “aging process,” and the latter modulates disease occurrence and severity. In other words, age-associated changes in cardiovascular structure and function become “partners” with pathophysiologic disease mechanisms, lifestyle, and genetics in determining the threshold, severity, prognosis, and therapeutic response of cardiovascular disease in older persons.
Figure 102–1.
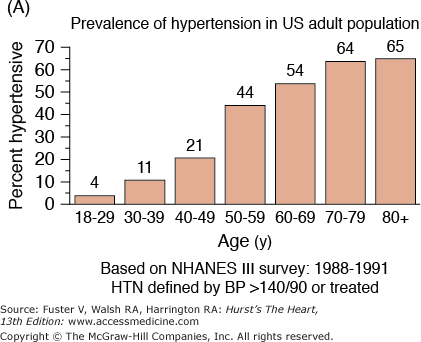
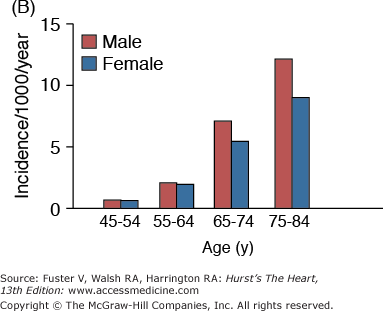
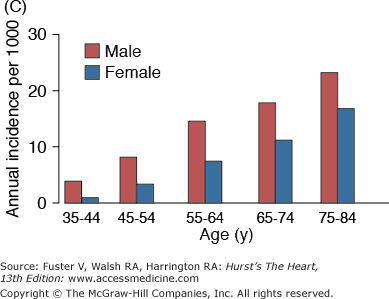
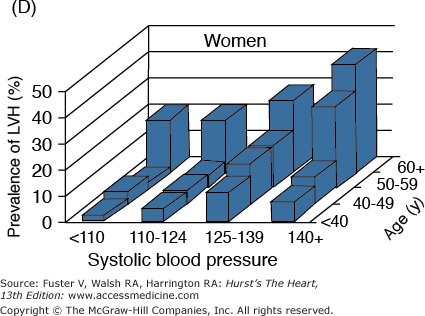

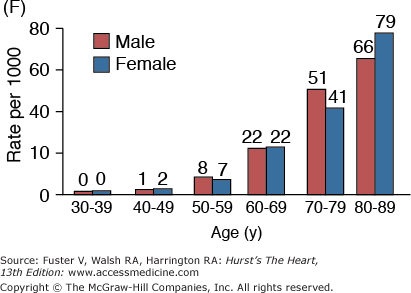

A. Prevalence of hypertension, defined as systolic blood pressure ≥140 mm Hg or diastolic blood pressure ≥90 mm Hg or current use of medication for purposes of treating high blood pressure. Data are based on National Health and Nutrition Examination Survey (NHANES) III (1988-1991). B. Incidence of atherothrombotic stroke (per 1000 subjects per year) by age in men and women from the Framingham Heart Study. C. Incidence of coronary heart disease by age in men and women from the Framingham Heart Study. D. Prevalence of echocardiographic left ventricular hypertrophy (LVH) in women according to baseline age and systolic blood pressure. E. Prevalence of echocardiographic LVH in men according to baseline age and systolic blood pressure. F. Prevalence of heart failure by age in men and women from the Framingham Heart Study. G. Prevalence of atrial fibrillation (AF) by age in subjects from the Framingham Heart Study. A to C, data compiled from Burt VL, Whelton P, Roccella EJ, et al. Prevalence of hypertension in the US adult population: results from the Third National Health and Nutrition Examination Survey, 1988-1991. Hypertension. 1995;25:305-313); Wolf PA. Lewis A. Conner lecture: contributions of epidemiology to the prevention of stroke. Circulation. 1993;88:2471-24783; Kannel WB, Wolf PA, Garrison RJ, eds. Framingham Study: An Epidemiological Investigation of Cardiovascular Disease. Section 34. NIH Publication No.87-2703. Bethesda, MD: National Heart, Lung and Blood Institute; 1987. D to G, data compiled from Levy D, Anderson KM, Savage DD, et al. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors: the Framingham Heart Study. Ann Intern Med. 1988;108:7-13; Ho KK, Pinsky JL, Kannel WB, et al. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. 1993;22:6A-13A; Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham Study. Stroke. 1991;22:983-988.
The nature of age-disease interactions is complex and involves mechanisms of aging, multiple defined disease risk factors, and as yet undefined risk factors. The role of specific age-associated changes in cardiovascular structure and function in such age-disease interactions has not been considered in most epidemiologic studies of cardiovascular disease.
Quantitative information on age-associated alterations in cardiovascular structure and function in health is essential to unravel age-disease interactions and to target the specific characteristics of cardiovascular aging that render it such a major risk factor for cardiovascular diseases. Such information is also of practical value to differentiate between the limitations of an older person that relate to disease and those that might be expected, within limits, to accompany advancing age or a sedentary lifestyle. During the past 3 decades, a sustained effort has been applied to characterize the effects of aging in health on multiple aspects of cardiovascular structure and function in a single study population, the Baltimore Longitudinal Study on Aging (BLSA). These community-dwelling volunteers are rigorously screened to detect both clinical and occult cardiovascular disease and are characterized with respect to lifestyle (eg, exercise habits) in an attempt to clarify the interactions of disease, risk factors, and aging itself. Perspectives gleaned from these studies are emphasized throughout this chapter.
Successful versus Unsuccessful Cardiovascular Aging
To define why age (or an aging process × exposure time) is so risky, the specific components of the risk associated with age must be identified. Two complementary approaches have evolved. Epidemiologists search for novel measures of “subclinical disease” (in addition to the more established risk factors that are already well characterized) in large, unselected cohorts composed of persons both with and without cardiovascular disease. In contrast, gerontologists attempt to develop quantitative information on cardiovascular structure and function in apparently healthy individuals to define the specific characteristics of aging that render it such a major risk factor for cardiovascular disease, even in the absence of clinically apparent comorbidity. The latter approach consists of identifying and selecting community-dwelling individuals who do not have (or have not yet experienced) clinical disease and who do not have occult disease that can be detected by noninvasive methods. These individuals are then grouped by age and stratified according to the level of a given variable, which may include some of the novel measures of subclinical disease identified by the epidemiologists. If the variable is perceived as beneficial or deleterious with respect to cardiovascular structure or function, those with extreme measures are considered to be aging “successfully” or “unsuccessfully,” respectively. Unsuccessful aging in this context is not synonymous with having clinical disease, because individuals with defined overt or occult clinical disease have been excluded from consideration a priori. Instead, unsuccessful aging, that is, falling within the poorest category with respect to the measure viewed as deleterious, may be viewed as a risk factor for future clinical cardiovascular disease. In this regard, unsuccessful aging is a manifestation of the interaction of the cardiovascular aging process and specific aspects of vascular disease pathophysiology. Thus, gerontologists and epidemiologists have become part of a joint effort in the quest to define why aging confers enormous risk for cardiovascular disease.
Age-Associated Changes in Arterial Structure and Function
Close examination of the age-associated changes in arterial structure and function may help explain why aging is such a strong predictor of adverse events. Findings from clinical studies show that the age-associated changes in arterial structure and function, previously not defined as clinical or subclinical diseases, are themselves risk factors for cardiovascular diseases. These novel risk factors, including intimal-medial thickness, arterial stiffness, and endothelial dysfunction, alter the substrate upon which the cardiovascular diseases are superimposed; therefore, they affect the development, manifestation, severity, and prognosis of these diseases.
Many age-associated changes are seen in the large arteries of humans. Cross-sectional studies show that central elastic arteries dilate with age, leading to an increase in lumen size5 (Fig. 102–2A), which results in increased inertance. In addition, postmortem studies have indicated an age-associated increase in arterial wall thickening, which is caused mainly by an increase in intimal thickening. In cross-sectional studies, carotid intimal-medial thickening increases nearly three-fold between the ages of 20 and 90 years6 (Fig. 102–2B). Note in Fig. 102–2B that both the average and range of intimal-medial thickness values are greater at higher ages, suggesting heterogeneity in the magnitude of the age-associated thickening process among older individuals. The increase in arterial wall thickening is accompanied by an increase in arterial stiffening (reduction in compliance)7 (Fig. 102–2C), which is due to several structural changes in the arterial wall.8 These changes include an increase in collagen content, cross-linking of adjacent collagen molecules to form advanced glycation end products, fraying of elastin, a decrease in the amount of elastin, and deposition of calcium in the medial layer. In addition to structural changes, functional alterations include an age-associated deterioration in vascular endothelial vasoreactivity.9
Figure 102–2.
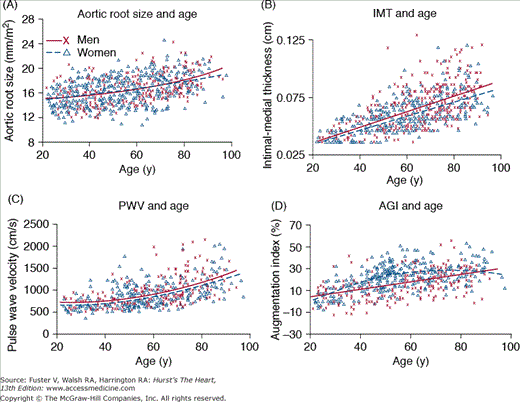
Age-associated changes in arterial structure and function in healthy Baltimore Longitudinal Study of Aging volunteer men (x) and women (D). Best fit regression lines (quadratic or linear) are shown for men (solid lines) and women (dotted lines). A. Aortic root size, measured via M-mode echocardiography. B. Common carotid intimal-medial thickness (IMT) as a function of age and sex. Note that the range of values for IMT is much greater in older individuals than in younger ones. C. Carotid-femoral pulse wave velocity (PWV). D. Carotid arterial augmentation index (AGI), which is defined as the ratio of the distance from the inflection point to the peak of the arterial waveform, over the pulse pressure. Note that unlike PWV, which increases quadratically with age, the age-associated increase in AGI is linear in men and convex shape in women, suggesting that factors other than stiffness also modulate the origin of reflected waves and the amplitude of AGI. Reproduced with permission from Najjar SS, Scuteri A, Lakatta EG. Arterial aging: is it an immutable cardiovascular risk factor? Hypertension. 2005;46:454-462.
Both systolic and pulse pressures increase with age in all adults, whereas diastolic blood pressure increases until the fifth decade of life and then levels off before decreasing after 60 years of age (Fig. 102–3). These age-dependent changes in systolic, diastolic, and pulse pressures are consistent with the idea that in younger people, blood pressure is determined largely by peripheral vascular resistance, whereas in older people, blood pressure is determined mainly by the stiffness of central conduit vessels.8
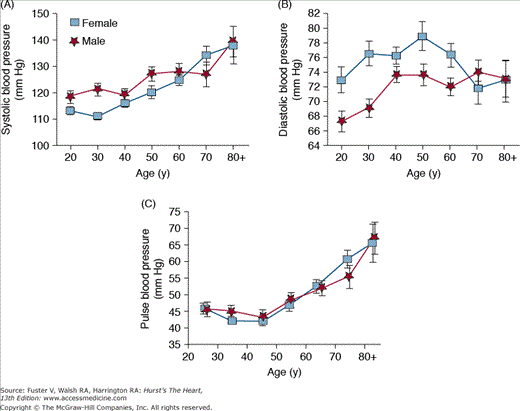
In older individuals, isolated systolic hypertension is the most common form of hypertension. Isolated systolic hypertension is defined as a systolic blood pressure >140 mm Hg and a diastolic blood pressure <90 mm Hg (ie, a widened pulse pressure), and it could be described as a disease related, in part, to arterial stiffening. Even mild isolated systolic hypertension (stage 1) is associated with an appreciable increase in cardiovascular disease risk10 and is an indication for treatment. Although the initial cutoff value for normal systolic pressure was 160 mm Hg, the value was adjusted downward when studies showed that pressures between 140 and 160 mm Hg conferred added risk. The systolic value was recently pushed further down to 130 mm Hg for patients with diabetes mellitus.11
Systolic blood pressure and pulse pressure in the central aorta may differ from the values that are measured more distally along the arterial tree due to the phenomenon of blood pressure amplification across the arterial tree.12 Even though peripheral systolic and pulse pressure increase with age, the central to peripheral pressure amplification decreases with age. Recent studies have shown that central blood pressures are independent predictors of outcomes13 and may even be better predictors of cardiovascular mortality than peripheral blood pressures.14 The usefulness and feasibility of integrating measurements of central arterial blood pressures into routine medical practice are areas of intense ongoing research.
Studies of morphologic, cellular, enzymatic, and biochemical changes in animal models have increased our understanding of age-associated arterial remodeling in humans. For example, the age-associated intimal-medial thickening seen in humans is often ascribed to “subclinical” atherosclerosis.15 This idea has become so well accepted that intimal-medial thickening is used by some investigators as a surrogate measure of atherosclerosis. However, intimal-medial thickening, which is usually measured in areas devoid of atherosclerotic plaque, is only weakly associated with the extent and severity of coronary artery disease.16 Furthermore, findings in rodent4 and nonhuman primate models of aging clearly indicate that intimal-medial thickening is an age-related process that is separate from atherosclerosis because atherosclerosis is absent in both of these animal models. Thus, excessive intimal-medial thickening is not necessarily synonymous with early or subclinical atherosclerosis.
Nonetheless, an association between intimal-medial thickening and atherosclerosis17 has been documented in humans. In individuals rigorously screened for the absence of clinical cardiovascular disease, excessive intimal-medial thickening at a given age predicts silent coronary artery disease6 (Fig. 102–4A), which, in turn, progresses to symptomatic ischemic heart disease. In the Atherosclerosis Risk in Communities (ARIC) study, which comprised middle-aged adults, intimal-medial thickening was associated with a greater prevalence of cardiovascular diseases and was an independent predictor of stroke.18 In the Cardiovascular Health Study (CHS), which comprised individuals older than age 65 years, intimal-medial thickening was an independent predictor of future myocardial infarction and stroke.19 In the CHS study, subjects were grouped according to quintiles of intimal-medial thickening, and the results indicated a nonlinear gradation in risk, with higher quintiles conferring a greater risk for cardiovascular diseases (Fig. 102–4B). Compared with the lowest quintile, the fifth quintile had a 3.15 relative risk for cardiovascular events, even after adjusting for traditional risk factors. In fact, the strength of intimal-medial thickening as a risk factor for cardiovascular diseases equals or exceeds that of most other traditional risk factors (Fig. 102–4C).
Figure 102–4.

Carotid intimal-medial thickness (IMT) and cardiovascular diseases. A. Common carotid artery IMT (CCA-IMT) as a function of age, stratified by coronary artery disease (CAD) classification, in Baltimore Longitudinal Study of Aging subjects. CAD-1 denotes a subset with positive exercise electrocardiogram (ECG) but negative thallium scans; CAD-2 represents a subset with concordant positive exercise ECG and thallium scans. B. Common carotid IMT as a predictor of future cardiovascular events in the Cardiovascular Health Study (CHS). Note the nonlinear increase in the risk for cardiovascular event rates with increasing quintiles. C. Comparisons of the associations of age- and sex-adjusted cardiovascular risk factors with the combined events of stroke or myocardial infarction in the CHS study, using Cox proportional hazards models. Note that IMT is a potent risk factor for future cardiovascular events. 1SD, one standard deviation; AF, atrial fibrillation; CV, cardiovascular; DBP, diastolic blood pressure; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SBP, systolic blood pressure. B and C, reproduced with permission from O’Leary et al.19
Thus intimal-medial thickening is not a manifestation of atherosclerosis but is associated with it. Intimal-medial thickening is an aging-related process that is separate from the pathophysiologic process of atherosclerosis, yet intimal thickening is a risk factor for atherosclerosis.
Studying the relationship between primary age-associated vascular wall remodeling and cardiovascular diseases has led researchers to search for new phenotypic manifestations of arterial remodeling to explore their clinical and prognostic significance. For example, the various carotid geometric patterns that are derived by combining the measurements of vascular mass with wall-to-lumen ratio were recently associated with unique functional and hemodynamic profiles that are largely independent of age and hypertension.20 These patterns were recently found to have differing prognostic implications.21
In addition to intimal-medial thickening, increased arterial stiffness has been observed with advancing age in humans and in animal models of aging.4 Strictly speaking, stiffness and its inverse, distensibility, depend on intrinsic structural properties of the blood vessel wall that relate a change in pressure with a corresponding change in volume. However, in this chapter, the terms stiffness and compliance are used in a broader sense to denote the overall lumped stiffness and compliance, which include the additional effects of vascular tone, blood pressure, and other modulating factors, all of which impact left ventricular (LV) afterload. Of note, in contrast to central arteries, the stiffness of muscular arteries does not increase with advancing age. Thus, the manifestations of arterial aging may vary among the different vascular beds, reflecting differences in the structural compositions of the arteries and, perhaps, differences in the age-associated signaling cascades that modulate the arterial properties or differences in the response to these signals across the arterial tree.
With each systolic contraction of the ventricle, a propagation wave that is generated in the arterial wall travels down the arterial tree. This propagation wave accompanies (and slightly precedes) the luminal flow wave generated during systole. The velocity of propagation of this wave is determined by the intrinsic stress-strain relationship of the vascular wall and by the smooth muscle tone, reflected by the mean arterial pressure.
The availability of noninvasive measures of the velocity of this pulse wave allows for large-scale epidemiologic studies. Pulse wave velocity was assessed in BLSA participants who were rigorously screened for the absence of overt or silent cardiovascular disease7 and in other populations with varying degrees of prevalence of cardiovascular disease.22 In all these studies, a significant age-associated increase in pulse wave velocity has been observed in both men and women.
Several clinical studies have recently shown the adverse cardiovascular effects of accelerated vascular stiffening. In the ARIC study, several indices of arterial compliance were predictors of hypertension.23 In the BLSA, pulse wave velocity was an independent predictor of incident hypertension.24 In hypertensive patients, pulse wave velocity was a marker of cardiovascular risk25 and coronary events26 and was an independent predictor of mortality.27 In addition, pulse wave velocity was an independent predictor of mortality in population-based studies,28,29 in subjects older than 70 years of age30 and in patients with end-stage renal disease31 (Fig. 102–5A). Other noninvasive indices of vascular compliance, including stroke volume divided by pulse pressure32 and the incremental modulus of elasticity,33 are also independent predictors of adverse outcomes. Thus, arterial stiffening, like intimal-medial thickening, should be viewed as another marker of aging, which, when accelerated, also becomes a risk factor for cardiovascular diseases.
Figure 102–5.
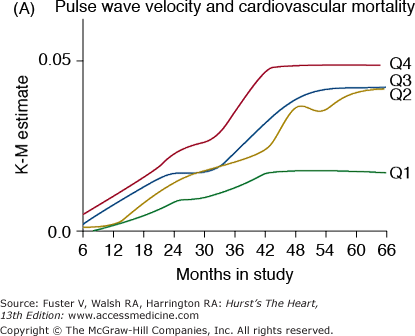
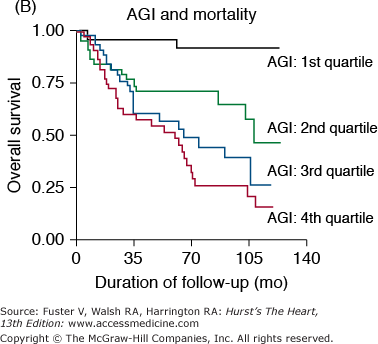
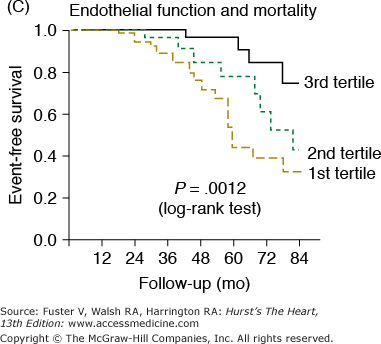
Markers of arterial aging are risk factors for adverse cardiovascular outcomes. A. Pulse wave velocity is a predictor of cardiovascular mortality in community-dwelling older subjects. This association remained significant after adjusting for age, sex, race, systolic blood pressure, known cardiovascular disease, and other variables related to events. K-M, Kaplan-Meier; Q, quartile. B. Probability of overall survival in patients with end-stage renal failure, stratified by quartiles of augmentation index (AGI). C. Probability of event-free survival in never treated hypertensive patients, stratified by tertiles of endothelial dysfunction. A, reproduced with permission from Sutton-Tyrrell et al.30B, reproduced with permission from London et al.39C, from Perticone F, Ceravolo R, Pujia A, et al. Prognostic significance of endothelial dysfunction in hypertensive patients. Circulation. 2001;104:191-196.
The interaction between vascular wall stiffening and cardiovascular diseases may set in motion a vicious cycle. Pulse wave velocity is determined, in part, by smooth muscle cell tone, which, in turn, is partially regulated by endothelial cells. Moreover, endothelial dysfunction occurs early in several cardiovascular disorders including atherosclerosis, diabetes, and hypertension. Thus, in this cycle, alterations in the mechanical properties of the vessel wall contribute to endothelial cell dysfunction and, ultimately, vascular stiffening.
In addition to the forward pulse wave, each cardiac cycle generates a reflected wave, which travels back up the arterial tree toward the central aorta. This reflected wave, which probably originates in the smaller arteries and arterioles, alters the arterial pressure waveform and is modulated, in part, by nitric oxide.34 The velocity of the reflected flow wave is proportional to the stiffness of the arterial wall. Thus, in young individuals whose vascular wall is compliant, the reflected wave does not reach the large elastic arteries until diastole. With advancing age and increasing vascular stiffening, the velocity of the reflected wave increases, and the wave reaches the central circulation earlier in the cardiac cycle, during the systolic phase.
This reflected wave can be noninvasively assessed from recordings of the carotid35 or radial36 arterial pulse waveforms by arterial applanation tonometry and high-fidelity micromanometer probes. Inspection of the recorded arterial pulse wave contour shows an inflection point, which heralds the arrival of the reflected wave. The difference in pressures between those at the inflection point and at the peak of the arterial waveform is the pressure pulse augmentation that is due to the early arrival of the reflected wave. Dividing this augmentation by the distance from the peak to the trough of the arterial waveform (corresponding to the pulse pressure) yields the augmentation index. The augmentation index, like the pulse wave velocity, increases with age in early and mid-adulthood7,15,35 (Fig. 102–2D). However, unlike pulse wave velocity, the rate of increase in the augmentation index decreases at older ages, particularly in women.37
Because reflected waves originate in small arteries and arterioles, the age-associated changes in this index are also probably determined, in part, by the age-associated changes in the structure and function of distal vessels and by age-associated alterations in the structure and function of large elastic arteries. Although attention has focused on the transmission velocity of reflected waves as an index of arterial stiffness, evaluation of the pulse wave contour may provide valuable insight into the characteristics and the pathology of more distal vessels, where reflected waves originate.38
The pressure pulse augmentation provided by the early return of the reflected wave is an added load against which the ventricle must contract. Furthermore, the loss of the diastolic augmentation present in compliant vessels caused by the late return of the reflected waves decreases diastolic blood pressure and thus has the potential to reduce coronary blood flow because most coronary flow occurs during diastole. These considerations suggest that excessively early return of the reflected waves may be detrimental to the cardiovascular system. In fact, the augmentation index has been shown to be a predictor of adverse events in end-stage renal disease patients39 (Fig. 102–5B). Thus, this index is another marker of vascular aging that is a risk factor for cardiovascular diseases.
The combination of arterial wall stiffening and early return of the reflected waves widens the pulse pressure. Age-associated decreased elasticity also increases the systolic pressure for any given volume of ejected blood and lowers diastolic pressure by diminishing elastic recoil. Thus, pulse pressure is a useful hemodynamic marker of the vascular stiffness of conduit arteries. Clinical and epidemiologic studies in several different populations with varying prevalences of cardiovascular diseases have confirmed the prognostic importance of pulse pressure.40 Furthermore, in several studies, pulse pressure was a stronger predictor of outcome than were systolic or diastolic blood pressures. This suggests the need for studies to evaluate whether pulse pressure should replace systolic or diastolic pressures as a screening criterion or as a therapeutic end point in the treatment of hypertension.
Recent studies showing that increased vascular stiffness may precede the development of hypertension have underscored the relationship between hypertension and arterial wall stiffening.41 An increase in mean arterial pressure (or peripheral resistance) can lead to a secondary increase in large-artery stiffness; however, the primary age-associated increase in large-artery stiffness can lead to an increase in arterial pressures. Thus, hypertension can be defined as a disease that is, in part, determined or modulated by properties of the arterial wall.
Endothelial cells are extremely important and powerful regulators of the vasculature (see Chap. 8). Several cardiovascular conditions and risk factors are associated with endothelial dysfunction, including hypercholesterolemia, insulin resistance, cigarette smoking, and heart failure. Endothelial cell dysfunction contributes to the pathogenesis of hypertension and atherosclerosis.42 In addition, endothelial cells play a pivotal role in regulating several arterial properties, including vascular tone, vascular permeability, angiogenesis, and the response to inflammation. Several features of these arterial properties undergo age-associated alterations in function. Endothelial-derived substances (eg, nitric oxide [NO] and endothelin-1) are determinants of large-artery compliance,43 suggesting that endothelial cells may modulate arterial stiffness. Endothelial function in central arteries, however, has not been directly assessed in humans. In the brachial artery, endothelial function, as assessed by agonist- or flow-mediated vasoreactivity, declines with advancing age.9 Several studies have demonstrated that impaired endothelial vasoreactivity, in both the coronary and peripheral arterial beds, is an independent predictor of future cardiovascular events (Fig. 102–5C).
With advancing age, NO-dependent mechanical and agonist-mediated endothelial vasodilatation is reduced in humans and animals. This vasoreactivity depends on NO generated by endothelial nitric oxide synthase (eNOS). In aging rats,44 activity of the eNOS isoform is markedly reduced. In addition, the bioavailability of NO may be reduced due to age-associated increases in the amounts of superoxide and nitrated tyrosine residues of proteins.45
Aging is associated with increased expression of adhesion molecules in rats and increased adherence of monocytes to the endothelial surface in rabbits.4 Adhesion molecules on the luminal surface of endothelial cells mediate leukocyte binding to endothelial cells and subendothelial migration. This process is probably facilitated by the actions of matrix metalloproteinases (MMPs).46 Serum levels of adhesion molecules show age-associated alterations in humans.47 In patients with hypercholesterolemia and ischemic heart disease, serum levels of soluble vascular cell adhesion molecule-1, but not soluble intercellular adhesion molecule-1 (ICAM-1), are positively associated with aging.48
In rat aortas, aging is associated with increased permeability to albumin.4 Moreover, glycosaminoglycans, which help regulate several arterial properties including vascular permeability, accumulate in greater number in the intima of older rabbits. Within hours of an acute arterial balloon injury to the rabbit carotid artery, the pericellular distribution of glycosaminoglycans is significantly reduced in the arterial wall, and this loss is associated with a significant expansion of the extracellular space. The glycosaminoglycans are rapidly replaced in the media but not in the developing neointima by smooth muscle cells.49
Telomeres are specialized DNA-protein complexes that form the ends of chromosomes, and they have been proposed as possible indicators of biologic, as opposed to chronologic, aging. Telomeres shorten with each replicative cell division, unless they are rescued by the enzyme telomerase reverse transcriptase. When telomere length reaches a critical size, reflecting numerous cycles of attrition, no further cellular replication is possible, and the cell becomes senescent. Telomere length has been shown to be inversely associated with atherosclerotic grade50 and with chronologic age in endothelial cells from human abdominal aorta, iliac arteries, and iliac veins.51 In a study of Danish twins, telomere length of chromosomes in white blood cells was negatively associated with pulse pressure.52 In a normotensive French cohort, telomere length of chromosomes in white blood cells was longer in women than in men but was associated with variations in pulse pressure and pulse wave velocity only in men.53 Loss of telomere function induces endothelial dysfunction in vascular endothelial cells, whereas inhibition of telomere shortening suppresses age-associated dysfunction in these cells.54 The impact of telomere-induced vascular senescence may be accentuated in older individuals, in whom studies indicate that the number55 and activity56 of progenitor cells is reduced, suggesting an age-associated diminution in regenerative capacity, which may contribute to the age-associated impairment in angiogenesis.57
Endothelial cells play a pivotal role in angiogenesis, in which new vessels grow from the existing microvasculature. Angiogenesis requires the migration and proliferation of endothelial cells in response to cytokines. The age-associated impairment in angiogenesis is partly a result of changes in the levels of extracellular enzymes, matrix proteins, and growth factors that affect endothelial cell migration.
Arterial Aging in Cardiovascular Diseases
Although the aforementioned changes in arterial structure and function with aging were previously thought to be part of normative aging, this concept was challenged when data emerged showing that these changes are accelerated in the presence of cardiovascular diseases and, as noted earlier, that they are risk factors for cardiovascular morbidity and mortality.
Patients with hypertension exhibit greater carotid wall thickness,58 central arterial stiffness,59 and central pressure augmentation60 than do normotensive subjects, even after adjusting for age. They are thought to have higher central arterial diameters,61 although this is presently debated.62-64 Hypertensive individuals exhibit endothelial dysfunction, and the mechanisms underlying their endothelial dysfunction are similar to the ones that occur with normotensive aging, albeit they appear at an earlier age.65 The normotensive offspring of hypertensives also exhibit endothelial dysfunction,66 suggesting that endothelial dysfunction may precede the development of clinical hypertension. Among hypertensive men, shorter telomere length of circulating white blood cells is associated with greater arterial stiffness.53 The metabolic syndrome, which is quite prevalent among older individuals,67 is associated with elevated carotid arterial thickness and stiffness.68 Diabetics also exhibit higher carotid intimal-medial thickness than nondiabetics and accelerated progression of intimal-medial thickness.69 Even though their central arterial stiffness is increased,59 this is not accompanied by an increase in the central pressure augmentation.70 Diabetics also exhibit endothelial dysfunction,71 which is also present in their first-degree relatives with insulin resistance.72
The circulating white blood cells of insulin-dependent diabetics have shorter telomere lengths than those from normoglycemic controls or non–insulin-dependent diabetics.73 Patients with atherosclerosis have increased thickness6,17 and stiffness74 of their central arterial walls, greater central pressure augmentation,75 and shorter telomere lengths on their circulating white blood cells.76 They also exhibit endothelial dysfunction, which has been implicated in the pathogenesis of atherosclerosis and is one its earliest pathologic manifestations.
Age-Associated Changes in Cardiac Structure and Function in Persons Without a Heart Disease Diagnosis
Overall cardiovascular structure and function vary dramatically among older individuals.77 Identification of cardiovascular structural and functional changes that reflect an “aging process” per se is a formidable task. Neither functional differences among individuals in cross-sectional studies nor changes over time within a given individual in longitudinal studies are necessarily manifestations of an aging process. Rather, interactions among aging, disease, and lifestyle must be considered in interpreting age-associated changes in cardiovascular structure and function as measured in various studies.8
A unified interpretation of identified cardiac changes that accompany advancing age, in otherwise healthy persons, suggests that, at least in part, these are adaptive, occurring to some extent in response to arterial changes that occur with aging1 (Fig. 102–6A). Central arterial stiffening and an increase in systolic and pulse pressures produce a late increased vascular impedance, moderate increase in LV wall tension, and increase in the thickness of the LV wall (Fig. 102–6A, B), largely due to an increase in ventricular myocyte size and to modest increases in collagen levels. Studies in large populations with broad age ranges demonstrate that arterial pressure, which varies directly with vascular loading, is a major determinant of LV mass and that the relative impact of age and arterial pressure on LV wall thickness varies with the manner in which study subjects are screened with respect to hypertension.78 The increase in LV wall thickness with aging reduces the expected increase in cardiac afterload caused by increased LV volume in older persons during stress79 (Table 102–1).
Figure 102–6.
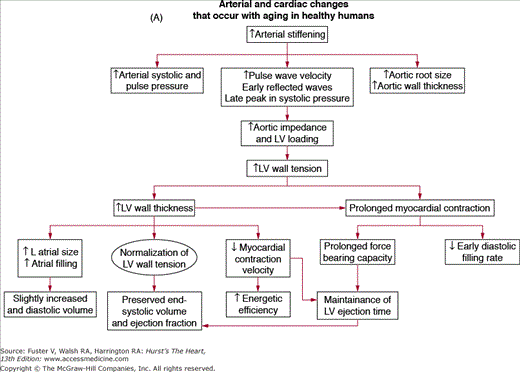
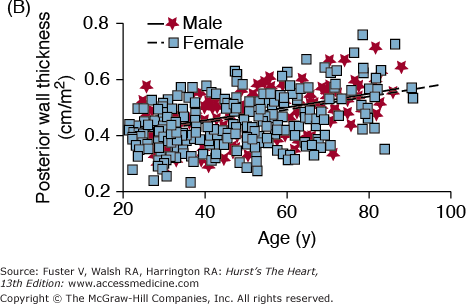
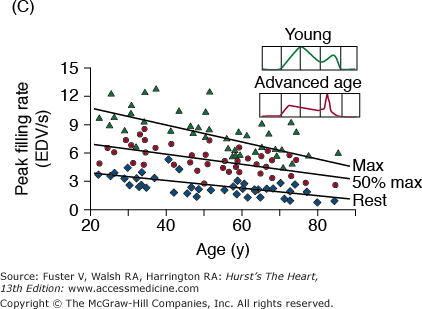
A. Arterial and cardiac changes that occur with aging in healthy humans. One interpretation (flow of arrows) of the constellation is that vascular changes lead to cardiac structural and functional alterations that maintain cardiac function. L, left; LV, left ventricular. B. Left ventricular posterior wall thickness, measured by M-mode echocardiography, increases with age in healthy Baltimore Longitudinal Study on Aging (BLSA) men and women. Note that the marked age-associated increase in left ventricular wall thickness in these healthy BLSA participants is within what is considered to be the clinically “normal” range. C. Maximum left ventricular filling rate at rest and during vigorous cycle exercise assessed via equilibrium gated blood-pool scans in healthy volunteers from the BLSA. EDV, end-diastolic volume. D. The ratio of early left ventricular diastolic filling rate (E) to the atrial filling component (A) declines with aging, and the extent of this E/A decline with aging in healthy BLSA volunteers is identical to that in participants of the Framingham Study. A, adapted from Lakatta EG. Normal changes of aging. In: Abrams WB, Berkow R, Rahway NJ. Merck Manual of Geriatrics. 2nd ed. Merck Sharp & Dohme, 1995. B, reproduced with permission from Gerstenblith et al.5C, reproduced with permission from Schulman SP, Lakatta EG, Fleg JL, et al. Age-related decline in left ventricular filling at rest and exercise. Am J Physiol. 1992;263:H1932-H1938. D reproduced with permission from Lakatta and Levy.104
| Oxygen consumption | ⇓ (50%) |
| (A-V)O2 | ⇓ (25%) |
| Cardiac index | ⇓ (25%) |
| Heart rate | ⇓ (25%) |
| Stroke volume | No ▵ |
| Preload | |
| EDV | ⇑ (30%) |
| Afterload | ⇑ |
| Vascular (PVR) | ⇑ (30%) |
| Cardiac (ESV) | ⇑ (275%) |
| Cardiac (EDV) | ⇑ (30%) |
| Contractility | ⇓ (60%) |
| Ejection fraction | ⇓ (15%) |
| Plasma catecholamines | ⇑ |
| Cardiac and vascular | ⇓ |
| Responses to β-adrenergic stimulation |
Prolonged contraction of the thickened LV wall maintains a normal ejection time in the presence of the late augmentation of aortic impedance. This preserves the systolic cardiac pumping function at rest. One disadvantage of prolonged contraction is that, at the time of the mitral valve opening, myocardial relaxation is relatively more incomplete in older than in younger individuals and causes the early LV filling rate to be reduced in older individuals (Fig. 102–6C). Structural changes and functional heterogeneity occurring within the LV with aging may also contribute to this reduction in peak LV filling rate. However, concomitant adaptations—left atrial enlargement and an enhanced atrial contribution to ventricular filling (Fig. 102–6D)—compensate for the reduced early filling and prevent a reduction of the end-diastolic volume. Age-associated changes in the tissue levels or responses (to growth factors, catecholamines, angiotensin II, endothelin, transforming growth factor β [TGFβ], or fibroblast growth factor) that influence myocardial or vascular cells or their extracellular matrices (see Lessons Learned About Cardiac Aging From Animal Models) may also have a role in the schema depicted in Fig. 102–6A.
Biologic sex is a well-recognized factor in the physiology and pathophysiology of the cardiovascular system, including the aging heart (reviewed in Leinwand80 and Konhilas and Leinwand81). Postmortem morphometric assessments in nonfailing human hearts have shown extensive age-related myocyte loss and hypertrophy of the surviving myocytes in males but preserved ventricular myocardial mass and average cell diameter and volume in aging female hearts.82 These sex differences may stem, in part, from differences in the replicative potential of cardiac myocytes. Analysis of gene expression differences by sex and age in LV samples from patients with dilated cardiomyopathy has identified more than 1800 genes displaying sexual dimorphism in the heart. A significant number of these genes were highly represented in gene ontology pathways involved in ion transport and G-protein–coupled receptor signaling.83
Impaired heart rate acceleration and impaired augmentation of blood ejection from the LV, accompanied by an acute modest increase in LV end-diastolic volume, are the most dramatic changes in cardiac reserve capacity that occur with aging in healthy, community-dwelling persons (see Table 102–1). Mechanisms that underlie the age-associated reduction in cardiovascular reserve are multifactorial and include (1) a reduction in intrinsic myocardial contractility; (2) an increase in vascular afterload (ventricular load is the opposition to myocardial contraction and the ejection of blood; afterload is the component of load that pertains to the time following excitation, as opposed to preload, prior to excitation); (3) arterial-ventricular load mismatching; (4) impaired autonomic regulation; and (5) physical deconditioning. Although these age-associated changes in cardiovascular reserve are insufficient to produce clinical heart failure, they do affect its clinical presentation—that is, the threshold for symptoms and signs or the severity and prognosis of heart failure secondary to any level of disease burden (eg, chronic hypertension that causes either systolic or diastolic heart failure).
Cardiac afterload has two components—one generated by the heart itself and the other by the vasculature. The cardiac component of afterload during exercise can be expected to increase slightly with age because the heart’s size increases in older persons throughout the cardiac cycle during exercise.79 The vascular load on the heart has four components: conduit artery compliance characteristics, reflected pulse waves, resistance, and inertance. Inertance is determined by the mass of blood in the large arteries that requires acceleration prior to LV ejection. As the central arterial diastolic diameter increases with aging (see Fig. 102–2A), the inertance component of afterload also likely increases. Thus, each of the pulsatile components of vascular load, measured at rest, increases with age. Hence, the aortic impedance, a composite function of the determinants of vascular afterload, increases with age.
Optimal and efficient ejection of blood from the heart occurs when ventricular and vascular loads are matched. (Note that in this context, stiffness refers to time-varying cardiac elastance throughout the cardiac cycle as a result of combined effects of active contractile and “passive” structural properties and to the interaction of both of these.) It has been suggested that the precise cardiac and vascular load matching that is characteristic in younger persons is preserved at older ages, at least at rest, because the increased vascular stiffness in older persons at rest is matched by increased resting ventricular stiffness.84
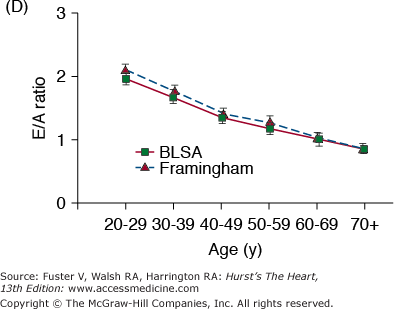
During exercise, in order for the ejection fraction to increase, the LV end-systolic elastance (ELV), that is, end-systolic pressure–to–end-systolic volume ratio, must increase to a greater extent than the effective vascular elastance (EA), that is, end-systolic pressure–to–stroke volume ratio. With increasing age, however, ELV fails to increase in proportion to the increase in EA; hence, the EA/ELV during exercise in older persons decreases to a lesser extent than it does in younger persons85 (Fig. 102–7A). This altered arterial-ventricular load matching in older versus younger persons during exercise is a mechanism for the deficit in the acute LV ejection fraction (LVEF) reserve that accompanies advancing age in many individuals. Thus, the LVEF, often attributed by cardiologists to a measure of LV pump function, is in fact determined by both cardiac and vascular properties, and both change with age. An acute pharmacologic reduction in both cardiac and vascular components of LV afterload by sodium nitroprusside (SNP) infusions in older, healthy BLSA volunteers augments LVEF (Fig. 102–7B) in these subjects (at rest and exercise).86 Because of concomitant reductions in preload and afterload during SNP infusion, the LV of older persons delivers the same stroke volume, stroke work, and cardiac output while working at a smaller size (Fig. 102–7C).
Figure 102–7.
A. Load mismatch during exercise. Effective arterial elastance index (EaI)/left ventricular end-systolic elastance (ELVI), an index of arterial-ventricular coupling, in men (dashed lines) and women (solid lines), <40 years of age (solid triangles) and >60 years of age, at rest, submaximal exercise (50% of max), and maximal exercise. In both men and women, EaI/ELVI decreases with exercise in both age groups, but there is a greater decrease in EaI/ElvI at maximal exercise in younger than in older subjects. B. Ejection fraction at seated, at upright rest, at intermediate common submaximal workloads, and at maximum effort in healthy volunteers aged 71 ± 7 years prior to and during sodium nitroprusside (SNP) infusion. At any level of effort, ejection fraction is substantially increased by SNP. C. Ventricular function, depicted as stroke work index versus end-diastolic volume index (EDVI) relationship at upright, at seated rest (R), and during exercise in the presence and absence of SNP. The relationship is shifted leftward and downward with SNP, indicating a smaller EDVI and lower stroke work index at any exercise load. A, reproduced with permission from Najjar et al.85B and C, reproduced with permission from Nussbacher et al.86
Diminished effectiveness of the autonomic modulation of heart rate, LV contractility, and arterial afterload collectively comprise a sizeable component of the age-associated deficit in cardiovascular reserve. The essence of sympathetic modulation of the cardiovascular system is to ensure that the heart beats faster; to ensure that it retains a small size, by reducing the diastolic filling period, reducing LV afterload; to augment myocardial contractility and relaxation; and to redistribute blood to working muscles and to skin, so as to dissipate heat. Each of the deficient components of cardiovascular regulation with aging (ie, heart rate [and thus filling time], afterload [both cardiac and vascular], myocardial contractility, and redistribution of blood flow) exhibits a deficient sympathetic modulation.
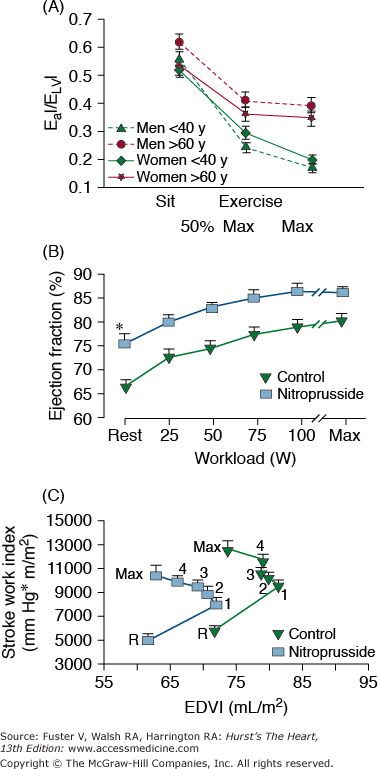
Multiple lines of evidence support the idea that the efficiency of postsynaptic β-adrenergic signaling declines with aging (see Lakatta1 for review). One line of evidence stems from the observations that cardiovascular responses to β-adrenergic agonist infusions at rest decrease with age (see Lakatta1 for review). A second type of evidence for a diminished efficacy of postsynaptic β-adrenergic receptor (AR) signaling is that acute β-AR blockade changes the exercise hemodynamic profile of younger persons to make it resemble that of older individuals. Significant β-blockade–induced LV dilatation occurs only in younger subjects (Fig. 102–8A). The heart rate reduction during exercise in the presence of acute β-adrenergic blockade is greater in younger versus older subjects (Fig. 102–8B), as are the age-associated deficits in LV early diastolic filling rate, both at rest and during exercise (Fig. 102–8C).
Figure 102–8.
A. Stroke volume index (SVI) as a function of end-diastolic volume index (EDVI) at seated rest (R) and during graded cycle workloads in the upright seated position in healthy Baltimore Longitudinal Study of Aging (BLSA) men in the presence and absence of β-adrenergic blockade. 1-5, graded submaximal workloads on cycle ergometer; max, maximum effort. Stroke volume versus end-diastolic functions with symbols are those measured in the presence of propranolol; dashed line functions without symbols are the stroke volume versus end-diastolic functions measured in the absence of propranolol. Note that in the absence of propranolol, the SVI versus EDVI relation in older persons (dashed brown lines) is shifted rightward from that in younger persons (dashed green lines). This indicates that the left ventricle (LV) of older persons in the sitting position compared with that of younger persons operates from a greater preload both at rest and during submaximal and maximal exercise. Propranolol markedly shifts the SVI-EDVI relationship in younger persons (▴) rightward, but does not markedly offset the curve in older persons (•). Thus, with respect to this assessment of ventricular function curve, β-adrenergic blockade with propranolol makes younger men appear like older ones. The abolition of the age-associated differences in the LV function curve after propranolol is accompanied by a reduction in heart rate, which at maximum effort is shown in B. B. Peak exercise heart rate in the same subjects as in A in the presence and absence of acute β-adrenergic blockade by propranolol. C. The age-associated reduction in peak LV diastolic filling rate at maximum exercise in healthy BLSA subjects is abolished during exercise in the presence of β-adrenergic blockade with propranolol. Y, subjects <40 years old; O, subjects >60 years old. A, reproduced with permission from Fleg JL, Schulman SP, O’Connor F, et al. Effects of acute beta-adrenergic receptor blockade on age-associated changes in cardiovascular performance during dynamic exercise. Circulation. 1994;90:2333-2341. B and C, data from Schulman SP, Lakatta EG, Fleg JL, et al. Age-related decline in left ventricular filling at rest and exercise. Am J Physiol. 1992;263:H1932-H1938.
Note, however, that β-adrenergic blockade in younger individuals in Fig. 102–8 causes the stroke volume index to increase to a greater extent than does β-adrenergic blockade in older individuals, suggesting that mechanisms other than deficient β-adrenergic regulation compromise LV ejection. One potential mechanism is an age-associated decrease in maximum intrinsic myocardial contractility. Another likely mechanism is enhanced vascular afterload caused by the structural changes in compliance arteries and possibly also by impaired vasorelaxation during exercise. In this regard, it has been observed that the increase in aortic impedance during exercise in old dogs is abolished by β-adrenergic blockade.87
Apparent deficits in sympathetic modulation of cardiac and arterial functions with aging occur in the presence of exaggerated neurotransmitter levels. Plasma levels of norepinephrine (NE) and epinephrine, during any perturbation from the supine basal state, increase to a greater extent in older compared with younger healthy humans (see Lakatta88 for review). The age-associated increase in plasma levels of NE results from an increased spillover into the circulation and, to a lesser extent, to reduced plasma clearance. The degree of NE spillover into the circulation differs among body organs; increased spillover occurs within the heart.89 Deficient NE reuptake at nerve endings is a primary mechanism for increased spillover during acute graded exercise. During prolonged exercise, however, diminished neurotransmitter reuptake might also be associated with depletion and reduced release and spillover. Cardiac muscarinic receptor density and function are also diminished with increasing age and might contribute to the decrease in baroreflex activity observed in aged subjects.90
Because a marked reduction in physical activity accompanies advancing age in a majority of adults,91,92 it may be hypothesized that a reduction in physical conditioning status might be implicated as a factor in the reduced cardiovascular reserve of older, healthy, sedentary individuals. The international standard for cardiorespiratory fitness is maximal oxygen uptake (VO2max). The extent to which the maximum aerobic capacity declines with aging and its suspected underlying mechanisms vary among studies. Aerobic capacity in persons of varying age, estimated by either peak oxygen consumption or work capacity, accompanying the hemodynamic pattern at maximal exercise across the age range (see Table 102–1) declines approximately 50% (Fig. 102–9A). In this study population, rigorously prescreened to exclude disease, the age-associated reduction in the cardiac component, exclusively a result of a reduced ability to accelerate the heart rate (Fig. 102–9B), accounts for roughly half of the age-associated decline in aerobic capacity, the remainder being attributable to age-associated differences in oxygen use. Such reductions in oxygen use during vigorous exercise result from age-associated reduction in muscle mass and from a reduction in the shunting of blood from viscera to working muscles during exercise, as well as the amount of oxygen use.
Figure 102–9.
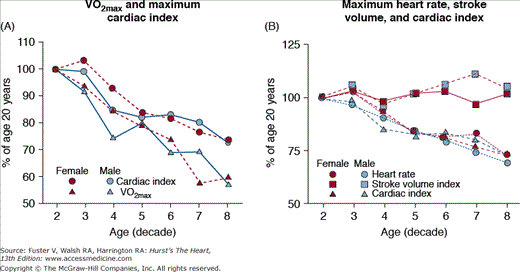
A. Peak oxygen consumption (VO2) and cardiac index at exhaustion during upright cycle exercise. The age-associated reduction in peak VO2 in both males and females is two-fold greater than the decline in maximum cardiac index. This indicates, via the Fick Principle, that oxygen extraction declines by approximately 25% over this age range. B. The age-associated decline in maximum cardiac index is entirely a result of a decline in maximum heart rate. Maximum stroke volume varies little with aging in healthy persons. VO2max, maximum oxygen consumption. Reproduced with permission from Fleg et al.79
Cross-sectional studies, for example, those in Fig. 102–9, have been largely interpreted to indicate that the VO2max declines linearly as a function of age. The longitudinal rate of decline in peak VO2 in healthy adults, however, is not constant across the age span in healthy persons, as assumed by cross-sectional studies, but accelerates markedly with each successive age decade, especially in men, regardless of physical activity habits (Fig. 102–10). When the components of peak VO2 were examined, the rate of longitudinal decline of the oxygen pulse (ie, the oxygen use per heart beat) mirrored that of peak VO2, whereas the longitudinal rate of heart rate decline averaged only 4% to 6% per 10 years and accelerated only minimally with age (see Fig. 102–10). The accelerated rate of decline of peak aerobic capacity has substantial implications with regard to functional independence and quality of life, not only in healthy older persons, but also when disease-related deficits are superimposed.
Figure 102–10.
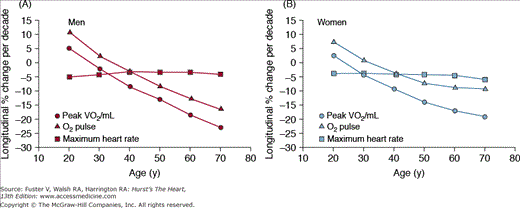
Longitudinal changes in maximal heart rate, oxygen (O2) pulse, and peak oxygen consumption (VO2) by sex, predicted from the mixed-effects model and separated by sex into panels A and B. Peak VO2/FFM (fat-free mass) declines progressively more steeply with advancing age, with similar declines in men and women. Note that peak VO2 per kilogram FFM is only slightly higher in men than women at younger ages, converging by old age. Declines in heart rate are similar across age in men but steepen modestly with age in women. O2 pulse declines progressively more steeply with age, especially in men, leading to near convergence of O2 pulse in elderly men and women. Note the similarity of these plots to those of peak VO2 in Fig. 102–1A. The longitudinal percent change per decade in maximal heart rate is only 4% to 5% per decade across the age span in both sexes. In contrast, longitudinal decline in O2 pulse accelerates progressively with age, especially in men. Note the similarity in the shape and magnitude of the decline in O2 pulse to that of peak VO2. Reproduced with permission from Fleg et al.92
Metabolic debts associated with the performance of dynamic exercise increase with aging. For several minutes after an acute bout of exercise, the body continues to consume oxygen at a rate in excess of the basal rate. This continued increased VO2 during recovery from exercise, sometimes referred to as an oxygen debt, when expressed relative to work performed during the exercise, can range from 14% to 21% of the total VO2 associated with the performance of and recovery from exercise.93 This oxygen debt, paid during recovery from exercise, often has not been factored into estimates of exercise efficiency (calories expended for work performed) and reduces efficiency because it contributes to the total oxygen cost associated with the exercise but contributes no external work.
Although work performed and VO2 during the exercise are lower in most older persons than in younger persons, the VO2 during the exercise, per unit work performed, does not change with age.93 VO2 during recovery (metabolic debt) from exercise in older subjects, however, exceeds that in younger persons by >30%. This debt, paid during recovery from exercise is likely attributable to an inability of the older body to adapt to the energy requirements of exercise. Specific factors include reductions in muscle mass and strength, inadequate blood flow to muscles, and a reduced efficiency of muscle respiration. Excessive elevation of catecholamines and core temperature occurs during exercise in older persons, as do other less well-characterized factors (eg, excessive reactive oxygen species and an exaggerated elevation of inflammatory cytokines). These factors cause muscle fatigue and a shift to anaerobic metabolism. During recovery, excessive catecholamine concentrations and incomplete waning of cell responses to catecholamine drive during exercise might continue to stimulate both muscle respiration in the absence of continued demand for muscle work and the cardiovascular system to dissipate lactate and heat generated during exercise. The physiologic significance of this metabolic debt incurred during exercise is that its underlying factors collectively reduce exercise capacity, with all the attendant health and performance drawbacks of such a reduction.
The issue arises as to whether physical conditioning via aerobic training of sedentary older persons can affect deficits in cardiovascular reserve capacity as a consequence of the aging process. It has been amply documented that physical conditioning of older persons can substantially increase their maximum aerobic work capacity and peak oxygen consumption. The extent to which this conditioning effect results from enhanced central cardiac performance or from augmented peripheral circulatory and oxygen use mechanisms, including changes in skeletal muscle mass, varies with the characteristics of the population studied, the type and degree of conditioning achieved, sex, body position during study, and likely genetic factors. A longitudinal study of older men in the upright position indicates that an enhanced physical conditioning status increases oxygen consumption and work capacity, in part, by increasing the maximum cardiac output by increasing the maximum stroke volume and, in part, by increasing the estimated total-body arteriovenous oxygen use.94 There is no strong evidence that physical conditioning of older persons can offset the deficiency in sympathetic modulation. Rather, conditioning effects to increase LV ejection appear to relate to the reduction in vascular afterload, as reflected in a reduced pulse wave velocity,95 and to carotid augmentation index in older athletes compared with sedentary controls, as well as possibly to an augmentation of the maximum intrinsic myocardial contractility.
Aerobic exercise training in older persons not only improves exercise work capacity but also increases muscle mass; improves capillary density, muscle respiration, mitochondrial enzymes, and muscle oxidative capacity; reduces plasma lactate during exercise; and reduces the oxygen debt. Thus, the good news is that the exercise training benefits on exercise efficiency of oxygen use are more pronounced in older than younger subjects, irrespective of sex. For example, in older subjects, an exercise conditioning program reduced the metabolic debt following exercise by nearly 30%, which translated into an 18% increase in exercise efficiency; but efficiency did not change in younger persons.93
There is an increase in elastic and collagenous tissue in all parts of the conduction system with advancing age. Fat accumulates around the sinoatrial node, sometimes producing a partial or complete separation of the node from the atrial musculature. There may be a pronounced decrease in the number of pacemaker cells in the sinoatrial node beginning at age 60 years, and by age 75 years, the sinoatrial node cell number may become substantially reduced. A variable degree of calcification of the left side of the cardiac skeleton, which includes the aortic and mitral annuli, the central fibrous body, and the summit of the interventricular septum, also occurs with aging. Because of their proximity to these structures, the atrioventricular (AV) node, AV bundle, bifurcation, and proximal left and right bundle branches may be affected by this process.
The PR interval increases with aging as a result of a prolongation of the A-H time with no change in the H-V time.96 Although the supine basal heart rate is not affected by aging, beat-to-beat fluctuation of heart rate, commonly known as heart rate variability, declines steadily with age.97 Reduced heart rate variability is an indicator of altered cardiac autonomic regulation commonly found in older people and has been linked to increased risk for morbid and fatal outcomes.97
An increase in the prevalence and complexity of both supraventricular and ventricular arrhythmias—whether detected by resting electrocardiogram (ECG), ambulatory monitoring, or exercise testing—occurs in otherwise healthy older, as opposed to younger, persons. Isolated atrial premature beats (APBs) appear on the resting ECG in 5% to 10% of subjects older than 60 years of age and are generally not associated with heart disease. Isolated APBs are detected in 6% of resting healthy BLSA volunteers older than 60 years of age, in 39% during exercise testing, and in 88% during ambulatory 24-hour monitoring.98 Over a 10-year mean follow-up period, isolated APBs, even if frequent, were not predictive of increased cardiac risk in these individuals.99
Short bursts of paroxysmal supraventricular tachycardia (PSVT) were observed in 1% to 2% of apparently healthy individuals older than 65 years of age who were rigorously screened to exclude disease. Several 24-hour ambulatory monitoring studies have demonstrated short runs of this PSVT (usually three to five beats) in 13% to 50% of clinically healthy older subjects.98,100 Although the presence of nonsustained PSVT did not predict an increase in risk of a future coronary event in BLSA subjects, 15% with PSVT later developed de novo atrial fibrillation (AF), compared with <1% of subjects without PSVT. The incidence of PSVT during exercise, typically asymptomatic three- to five-beat salvos, increases with age, from nil in the youngest age group to approximately 10% in the ninth decade.101 Although individuals with exercise-induced PSVT were not at a greater risk for coronary events over a multiyear follow-up, 10% developed a spontaneous atrial tachyarrhythmia, compared with only 2% of the control group. Thus, PSVT at rest or induced by exercise is an early clue that some healthy individuals are at increased risk for future AF. Another risk factor for AF may be the increase in left atrial size that accompanies advancing age in otherwise healthy persons.5
In older subjects without apparent heart disease, the limited data available support a marked age-associated increase in the prevalence and complexity of ventricular ectopy (VE), both at rest and during exercise, at least in men. A steep increase in the prevalence of VE with advancing age occurs both in those clinically free of heart disease and in unselected populations. In healthy BLSA volunteers with a normal ST-segment response to treadmill exercise, isolated VE occurred at rest in 8.6% of men older than age 60 years compared with only 0.5% in men 20 to 40 years of age. Interestingly, the prevalence of VE at rest was not age-related in women. Among 98 carefully screened asymptomatic BLSA participants older than 60 years of age, 35% had multiform isolated VE, 11% had ventricular couplets, and 4% had short runs of ventricular tachycardia on 24-hour monitoring98; all occurred substantially more commonly in older than in healthy younger persons. Neither the prevalence nor the complexity of resting VE was a determinant of future coronary events over a 10-year mean follow-up period.99 Isolated VE during or after maximal treadmill exercise increased in prevalence five-fold, from 11% to 57%, between the third and ninth decades in apparently healthy BLSA volunteers.102
Many of the age-associated changes in cardiac and arterial structure or function that have been observed in humans also occur across a wide range of other species. Insights gained from cellular and molecular studies in these animal models may hold clues that will assist in directing future efforts toward developing novel therapies for age-associated arterial structural and functional remodeling in humans. The results of studies have been reviewed.2,4,103
Lessons Learned About Cardiac Aging from Animal Models
Cellular and molecular mechanisms implicated in age-associated changes in myocardial structure and function in humans have been largely studied in rodents. Many experimental studies have shown that increased activation of the renin-angiotensin-aldosterone system is a prominent feature of age-related cardiac remodeling that may account for many of the phenotypic changes that are observed.8,104 The altered cardiac structural phenotype that evolves with aging in rodents includes an increase in LV mass due to an enlargement of myocyte size105 and focal proliferation of the matrix in which the myocytes reside, which may be linked to an altered cardiac fibroblast number or function. The number of cardiac myocytes becomes reduced because of necrosis and apoptosis, with the former predominating.106 Putative stimuli for cardiac cell enlargement with aging in rodents include an age-associated increase in vascular load due to arterial stiffening and stretching of cells caused by drop out of neighboring myocytes.107 Stretch of cardiac myocytes and fibroblasts initiates growth factor signaling (eg, angiotensin II/TGFβ), which, in addition to modulating cell growth and matrix production, leads to apoptosis.108 In mouse heart, activation of the calcineurin-NFAT pathway increases with age.109 The expression of atrial natriuretic110 and opioid111 peptides, molecules that are usually produced in response to chronic stress, is increased in the senescent rodent heart. There is evidence that transcriptional events associated with hypertrophic stressors become altered with advancing age (eg, the nuclear binding activity of the transcription factor nuclear factor-κB is increased and that of another transcription factor, Sp1, is diminished).112 A recent proteomic comparison study113 discovered 117 proteins to be differentially expressed in the aged LV. An increase in oxidative stress is also implicated in age-related cardiac remodeling,114,115 and aging is indeed well recognized to be associated with increased production of reactive oxygen species (ROS) in many different tissues.116
Coordinated changes in the function or expression of proteins that regulate several key steps of the cardiac cell excitation-contraction coupling process occur in the rodent heart with aging and result in a prolonged action potential (AP), a prolonged cytosolic calcium (Cai) transient after excitation, and a prolonged contraction (see Lakatta and Sollott103 for review). Both the apparent number and the activity of individual cardiac L-type Ca channels increase with age in the rat. The ensemble L-type current inactivates more slowly in myocytes from older rats versus those in younger rats, and this, as well as reductions in outwardly directed K+ currents, could account, in part, for the prolonged AP of the former. The imposition of a shorter AP to myocytes from the old rat heart reduces the Cai transient amplitude, an effect attributable to a reduction in sarcoplasmic reticulum Ca2+ load, which is presumably due to a reduced Ca2+ influx via L-type Ca2+ channel and likely also to an increased net Ca2+ extrusion via sarcolemmal Na+-Ca2+ exchanger.
The prolonged Cai that occurs with aging is explained, in part, by a reduction in the rate of Ca2+ sequestration by the sarcoplasmic reticulum. This is attributable to a reduced transcription of the gene coding for the sarcoplasmic reticulum Ca2+ pump, SERCA2 (see Lakatta and Sollott103 for review). The abundance of transcripts of the cardiac Na+-Ca2+ exchanger (NCX-1), which serves as the main transsarcolemmal Ca2+ extrusion mechanism, is increased approximately 50% in senescent (24-month) compared with the young adult (6-month) rat hearts. The age-associated prolonged Ca2+ transience and contraction may impair myocardial relaxation during early diastole and, in part, may underlie the reduction in early diastolic filling rate that accompanies advancing age. Studies in rodents that have used gene therapy or exercise conditioning to increase the level of SERCA2 have demonstrated that impaired relaxation and Ca2+ sequestration that occur with aging can be reduced by these interventions.
Marked shifts occur in the cardiac myosin heavy chain (MHC) isoforms in the senescent rat heart, with the β-isoform becoming predominant; myosin Ca2+-ATPase activity declines with the decline in α-MHC content. MHC gene expression is regulated, in part, via binding of thyroid hormone receptors, members of a superfamily of receptors that includes retinoic acid receptors, vitamin D receptors, and retinoid X receptors, to thyroid response elements in gene 5′ flanking regions. A significant reduction (∼50%) in the levels of thyroid hormone receptor-β and retinoid X receptor-γ proteins between 6 and 24 months and in mRNA encoding these receptor subtypes has been observed in hearts of senescent rats (see Lakatta and Sollott103 for review).
The altered composite cellular profile, which results in a contraction that exhibits reduced velocity and a prolonged time course due to prolonged elevation of cytosolic Ca after a prolonged AP, can be considered to be adaptive rather than degenerative because myocardial shortening at reduced velocity is energy efficient. This altered pattern of Ca2+ regulation and myosin protein expression allows the myocardium of older hearts to generate force and active stiffness for a longer time after excitation (see Lakatta and Sollott103 for review). The prolonged isovolumic relaxation period that occurs in the healthy human heart with aging may be in part attributable to such prolonged contractile protein Ca2+ activation, enabling the continued ejection of blood during late systole, a beneficial adaptation with respect to enhanced central arterial stiffness and early reflected pulse waves.
The age-associated reduction in β-AR signaling may, in part, also be related to upregulation of opioid peptide receptor signaling because of the significant antagonistic effects between stimulation of opioid peptide receptor and β-AR–mediated positive contractile response.117
Remarkably, the available examinations of cardiac excitation-contraction coupling in rodents of both sexes118 or females only119 show a lack of age-related changes in the configuration of the Ca2+i transient in myocytes from female hearts. The somewhat unexpected findings in aged female myocytes include (1) an apparent increase in the sarcoplasmic reticulum Ca2+ content, (2) the lack of the effect of the latter on the configuration of the Ca2+i transients and contraction, and (3) a reduction of fractional sarcoplasmic reticulum Ca2+ release in the presence of unchanged ICaL
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


