Acute Respiratory Distress Syndrome
Joseph LoCicero III
Although references to pulmonary dysfunction were made as early as World War I, there was little understanding of the mechanisms and course of this devastating disease. In 1925, Osler93 observed that “uncontrolled septicemia leads to a frothy pulmonary edema that resembles serum, not the sanguinous transudative edema fluid seen in dropsy or congestive heart failure.” Early observations of the association between pulmonary dysfunction and trauma were made by Burford and Burbank in 1945,16 when they reported acute respiratory failure (“traumatic wet lung”) after thoracic injury and noted its association with increased interstitial and intra-alveolar fluid. Mallory and colleagues71 subsequently described some of the classic features of pulmonary pathology in necropsy cases of traumatic shock from World War II. However, it was not until 1967 that Ashbaugh and associates6 definitively described postoperative respiratory distress in adults. Then in 1971, Petty and Ashbaugh95 called the dysfunction adult respiratory distress syndrome (ARDS). Over the next 10 to 20 years, significant advances in resuscitation and ventilatory support led to a greater recognition of the syndrome of respiratory failure in patients with sepsis, shock, aspiration pneumonia, pancreatitis, and severe traumatic injuries. A number of terms—including shock lung, wet lung, Da Nang lung, and postperfusion lung—were used to describe the pulmonary dysfunction that developed in these patients, but eventually ARDS became the accepted term.
Although ARDS is a relatively common clinical entity, its incidence and prevalence are not well established. The ARDS Network did an intensive review of the registry and of data from the American Hospital Association to estimate the incidence of acute lung injury in the United States; many such cases go on to ARDS. The findings of the network reported by Goss and colleagues43 in 2003 were that the incidence is much higher than previously thought. Acute lung injury currently occurs at a rate of 64.5 cases per 100,000 patients, or 64.5 cases per 105 patient-years.
Definition of Ards
Since the naming of ARDS, many studies have claimed varying success in changing the usual dismal outcomes. One of the reasons for discrepancy among studies was the lack of a clear definition of the syndrome. The American–European Consensus Conference (AECC) on ARDS, reported by Bernard and associates,10 took on the task of redefining the syndrome. They accommodated by addressing a broader spectrum of pulmonary dysfunction and defining two clinical entities: acute lung injury and ARDS (Table 75-1).
With the adoption of these definitions, a uniform standard is now present to characterize the natural history, pathogenesis, and management outcomes of ARDS. A prospective comparison by Moss and associates89 of the definition proposed by the AECC and two alternative definitions, the Lung Injury Severity Score (LISS), which is defined later, and the Modified Lung Injury Severity Score, found that similar patients were identified by all three definitions. The LISS and the AECC definitions were compared subsequently in a series of patients by a Canadian consortium reported by Meade and colleagues78 and showed reasonable correlation.
Risk Factors and Biological Markers
Clinical risk factors, physiologic responses, and biological markers help to identify patients at risk for ARDS. Both direct and indirect events that cause pulmonary injury are associated with the development of ARDS (Table 75-2). Bernard10 reported on the American-European conference on Respiratory Distress, which established the etiology of ARDS. Both direct lung injury such as pulmonary contusion, gastric aspiration, and pulmonary infection and indirect lung injury such as sepsis, severe trauma, and prolonged shock are the most common causes of ARDS.
Clinical Risk Factors
Fowler and associates36 noted that nearly 80% of patients who develop ARDS have one or more of these risk factors. They pointed out that the risk of developing ARDS ranges from 2% to 40% in patients with a single risk factor. In their study, the probability of developing ARDS is disproportionately increased when more than one risk factor is present. The percentage of patients who develop ARDS is increased 2.3-fold when two risk factors are present and 4.7-fold with three risk factors. These same authors, as well as Hudson and colleagues,63 note that patients with uncontrolled sepsis, pulmonary contusion, aspiration pneumonia, prolonged shock, or severe polytrauma are at greatest risk for ARDS.
Table 75-1 Acute Lung Injury and Acute Respiratory Distress Syndrome | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Physiologic Responses
ARDS risk also depends on the magnitude of the physiologic insult, as indicated by correlation with the criteria for systemic inflammatory response syndrome (SIRS), the Injury Severity Score (ISS), and the Acute Physiology And Chronic Health Evaluation (APACHE) score (Table 75-3). The systemic inflammatory response syndrome, an early nonspecific index of systemic inflammation according to Rangel-Frausto and associates,98 is associated with a modestly increased risk of ARDS. This risk increases with the number of SIRS criteria met, but only 2% to 6% of patients with SIRS develop ARDS. ARDS risk increases more significantly with increasing APACHE II and ISS scores. Webster and coworkers111 observed that 44% of patients with an APACHE II score >29 and 50% of patients with an ISS >49 develop ARDS.
Biological Markers
In hopes of early intervention, researchers have sought the “holy grail” of a simple blood test or a biochemical marker that will herald the development of ARDS. Unfortunately all of the markers appear after the development of acute lung injury. Identifying markers that are present early in ARDS has not been futile, since some of them are reasonable prognostic factors. In 2005, Ware110 reported that the following factors had been studied in single institution situations all with some prognostic value: von Willebrand factor antigen, alterations in circulating coagulation and fibrinolytic proteins, interleukin-8, hepatocyte growth factor, transforming growth factor-alpha, and many others.
Table 75-2 Clinical Risk Factors for Acute Respiratory Distress Syndrome | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
Collaborative investigators collected data on prognostic factors in patients enrolled in the protective ventilation trials coordinated through the ARDS Network of the National Institutes of Health (NIH). Biologic markers associated with mortality included elevated levels of cytokines, markers of endothelial and alveolar epithelial injury, and abnormal coagulation and fibrinolysis. McClintock and colleagues76 analyzed the pooled data and reported their findings in 2008. In patients receiving protective ventilation strategies, they looked at plasma biomarkers of inflammation (IL-6, IL-8, intercellular adhesion molecule–1 [ICAM-1]); coagulation (thrombomodulin, protein C); and fibrinolysis (plasminogen activator inhibitor–1). They found that all markers except IL-6 were significantly different between survivors and nonsurvivors (Table 75-4). Nonsurvivors had more abnormal values. Three biomarkers—IL-8, ICAM-1, and protein C—remained significantly different by multivariate analysis, which included age, gender, Simplified Acute Physiology Score II, and all biomarkers that were significant on bivariate
analysis. Higher levels of IL-8 and ICAM-1 were independently predictive of worse outcomes (odds ratio = 2.0 and 5.8, respectively; p = 0.04 for both). Lower levels of protein C were independently associated with an increased risk of death (odds ratio = 0.5), a result that nearly reached statistical significance (p = 0.06). Their message was that in a group of patients treated with strict low-tidal-volume ventilation, independent predictors of mortality included elevated plasma biomarkers for inflammation (IL-8) and for enhanced neutrophil recruitment (ICAM-1).
analysis. Higher levels of IL-8 and ICAM-1 were independently predictive of worse outcomes (odds ratio = 2.0 and 5.8, respectively; p = 0.04 for both). Lower levels of protein C were independently associated with an increased risk of death (odds ratio = 0.5), a result that nearly reached statistical significance (p = 0.06). Their message was that in a group of patients treated with strict low-tidal-volume ventilation, independent predictors of mortality included elevated plasma biomarkers for inflammation (IL-8) and for enhanced neutrophil recruitment (ICAM-1).
Table 75-3 Acute Respiratory Distress Syndrome Risk | ||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||
Table 75-4 Multivariate Logistic Regression for Clinical and Biological Predictors of Mortalitya | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
Pathogenesis
Theories of the pathogenesis of ARDS must accommodate three simple yet challenging observations. First, the characteristics of ARDS are remarkably consistent despite its diverse clinical causes. The earliest accounts of ARDS highlighted the similarity of lung injuries produced by such diverse insults as sepsis, shock, fat embolism, gastric aspiration, and pulmonary contusion. This suggested the presence of a final common pathway or pathways of lung injury that could be elicited by a heterogeneous group of primary stimuli. Second, inflammatory stimuli presenting on either side of the alveolar–capillary membrane produce the same syndrome of pulmonary dysfunction. Because both direct and indirect insults produce ARDS, the final common pathway involved in lung injury may be induced by either intravascular or intra-alveolar stimuli. Third, identical insults do not produce lung injury in all individuals. This last and most challenging observation requires that hypotheses involving the pathogenesis of ARDS explain the heterogeneity of individual responses to a given stimulus.
Initial clinical and histologic investigations by Jenkins66 and Ashbaugh6 and their colleagues suggested that the refractory hypoxemia characteristic of ARDS was directly related to increased pulmonary capillary permeability and extravascular lung fluid. Both intra-alveolar and interstitial edema are consistent early features of ARDS. But, as noted by Mitchell and associates,85 although extravascular lung water is increased from three to eight times normal, it is the cellular process that produces these changes. The presence of leukocyte and platelet microthrombi, according to Bachofen and Weibel,7 indicates that ARDS is associated with an acute intravascular inflammatory process (Table 75-4). In 2008, Hedenstierna and Lattuada53 reviewed the literature on lymphatics and lymphatic flow in acute lung injury suggesting a more important role for obstructive edema in the mechanism of acute lung injury and the development of ARDS.
Proinflammatory Events
Critical proinflammatory events associated with the development of ARDS include macrophage activation; neutrophil recruitment and activation; endothelial injury; platelet aggregation and degranulation; activation of plasma proteins; and alveolar epithelial injury (Table 75-5). The initial sequence of cellular events that precede ARDS depends on the nature and site of the insult. Well-recognized differences in the nature of insults activate distinct inflammatory pathways. Recognition of the importance of the site of the primary insult in determining early inflammatory responses has increased. For example, indirect pulmonary insults like sepsis, shock, and pancreatitis cause early events such as activation of plasma protein systems and upregulation of adhesion molecules, causing neutrophil adherence and activation. In contrast, insults presented via the tracheobronchial tree—like gastric aspiration, pneumonia, and inhalation burn—first produce a local inflammatory response mediated in large part by alveolar macrophages. Subsequent diffuse lung injury depends on a failure of regulatory mechanisms to contain or localize the inflammatory response. In this setting the participation of intravascular components of the inflammatory response, including neutrophil recruitment and activation, occurs as a secondary event. The failure of initial attempts to treat ARDS by modifying inflammatory responses is in large part due to an incomplete understanding of the complexity of the factors that dictate host responses to injury and infection.
Macrophage Activation
Pulmonary interstitial and alveolar macrophages are key signaling and effector cells in acute lung injury. These resident leukocytes of the lung are ideally positioned to initiate local inflammatory responses. They do so by elaborating a variety of secreted and membrane-associated mediators, including cytokines, reactive oxygen intermediates, eicosanoids, tissue factor, and HLA-DR antigen. Macrophage activation promotes beneficial effects
such as neutrophil recruitment, antigen presentation, phagocytosis, and sequestration at the inflammatory site. But this activation also causes the release of a number of macrophage products, including reactive oxygen intermediates, tumor necrosis factor (TNF), and IL-1, all of which are implicated in acute lung injury, as reported by Brackett and McCay.15 As shown by Frank and colleagues37 in 2006, these factors are now thought to be integral contributors to ventilator-associated lung injury.
such as neutrophil recruitment, antigen presentation, phagocytosis, and sequestration at the inflammatory site. But this activation also causes the release of a number of macrophage products, including reactive oxygen intermediates, tumor necrosis factor (TNF), and IL-1, all of which are implicated in acute lung injury, as reported by Brackett and McCay.15 As shown by Frank and colleagues37 in 2006, these factors are now thought to be integral contributors to ventilator-associated lung injury.
Table 75-5 Cellular Pathogenesis of Acute Respiratory Distress Syndrome | |
|---|---|
|
Neutrophil Recruitment and Activation
Neutrophil participation in ARDS is well established. The presence of activated neutrophils in the lungs of patients with ARDS induces lung injury. Simms and D’Amico105 reported that neutrophil margination and transendothelial migration are mediated by chemoattractants and the upregulation of neutrophil–endothelial binding. Donnelly23 and Hirani57 and their colleagues observed that the neutrophil chemoattractants IL-8, LTB4, and C5a are increased in acute lung injury. IL-8 concentrations have been shown by Goodman and coworkers42 to correlate with the number of neutrophils present in the bronchoalveolar lavage fluid of patients with ARDS. Expression of neutrophil and endothelial adhesion molecules is also increased in acute lung injury. Simms and D’Amico105 as well as O’Leary92 and their associates, reported that proinflammatory mediators—including TNF, IL-1, and endotoxin released during conditions that predispose to ARDS—upregulate neutrophil expression of a number of adhesion molecules, including CD11b/CD18, ICAM-1, and L-selectin. The role of selectins in attracting and trapping neutrophils by margination was outlined by Donnelly and colleagues.24
Shappell and colleagues104 have suggested that integrins, in addition to their role in neutrophil margination, may further contribute to lung injury by inducing the release of reactive oxygen species. Blockade of CD11/CD18 on neutrophils or ICAM-1 on endothelial cells with monoclonal antibodies, as noted by Horgan and associates,61 attenuates pulmonary neutrophil sequestration and lung injury. Weiland and coworkers113 observed that activated neutrophils produce reactive oxygen intermediates—elastase, gelatinases, and collagenase—which are all capable of producing lung injury. Donnelly and colleagues25 noted that elastase is also an activator of neutrophils.
Despite the evidence that neutrophils play an important role in the pathogenesis of ARDS, neutrophil-independent mechanisms also appear to be involved in lung injury. Acute lung injury has been demonstrated in severely neutropenic patients by Ognibene and colleagues91 and in animal models with impaired neutrophil adherence or attenuated neutrophil function by Carraway and associates.17
Endothelial Injury
Pulmonary endothelial injury is a hallmark of ARDS. Ashbaugh and colleagues6 initially stated that the earliest changes included alveolar hemorrhage, interstitial and intra-alveolar edema, microvascular occlusion with platelet and neutrophil aggregates, and loss of normal endothelial histocytologic architecture. Sabharwarl and associates100 showed that biochemical markers of endothelial cell activation and injury are elevated in patients with ARDS. Grau and coworkers44 noted that the activation of tumor necrosis factor (TNF) receptor p75, ICAM-1, and vascular cell adhesion molecule–1 (VCAM-1) is also increased in pulmonary microvascular endothelial cells from patients with ARDS.
Neutrophil adherence and the creation of a protected microenvironment between the neutrophil and endothelial cell are prerequisites for neutrophil-mediated injury to endothelium. Inflammatory mediators released during injury induce the expression of endothelial adhesion molecules. Chuluyan and colleagues19 noted that IL-1 upregulates E-selectin, ICAM-1, and VCAM-1 expression. Shalaby and associates103 showed that TNF and IL-1 enhance endothelial injury by neutrophils and reactive oxygen intermediates. Minamiya and colleagues84 observed that bacterial lipopolysaccharides also promote neutrophil–endothelial adherence and release of hydrogen peroxide into the pulmonary microcirculation.
Zimmerman and coworkers,120 in a 1999 review, summarized the findings that endothelial injury associated with neutrophil adherence and activation results in a loss of endothelial integrity characterized by cytoskeletal changes, increased permeability, and proteolytic injury to endothelial surface elements. Recovery of endothelial membrane–associated proteins from extracellular fluids is increased. In patients with ARDS, these include thrombomodulin and soluble E-selectin. Zimmerman and colleagues also noted that release of soluble E-selectin may further exacerbate endothelial injury by decreasing neutrophil chemotaxis and increasing the production of reactive oxygen intermediates and integrin-mediated adhesion.
In 2008, Maniatis and Orfanos72 summarized the most recent investigations into the molecular and cellular aspects of the pathogenesis of endothelial injury, suggesting interventions derived from this recent science which are yet to be evaluated.
Platelet Aggregation and Degranulation
Platelet aggregation and microvascular thrombosis normally act to sequester and limit inflammatory responses. In contrast, disseminated platelet activation contributes to pulmonary dysfunction by redistributing microvascular blood flow, increasing capillary permeability, and activating leukocytes. According to the studies of Heffner and coworkers,54 platelet activation may be initiated by bacterial lipopolysaccharides, platelet-activating factor, thromboxane, and thrombin. They reported that degranulation further amplifies the local inflammatory response, producing vasoconstriction and neutrophil recruitment by the release of platelet-activating factor, thromboxane, serotonin, and platelet-derived growth factor.
Activation of Plasma Protein Systems
Plasma protein systems are activated in ARDS. Craddock and colleagues20 observed complement activation commonly in ARDS; it may be used experimentally to produce lung injury. Complement fragments are potent activators of neutrophils, stimulating aggregation, chemokinesis, and adhesion to endothelium. However, both Flick33 and Dehring22 and their colleagues showed that depletion of complement does not prevent septic lung injury, suggesting that other mechanisms contribute.
A second plasma protein system commonly activated in ARDS is the coagulation cascade. Dorinsky and Gadek27 found that clinically evident coagulopathy is present in 26% of patients
with ARDS. Intravascular and intra-alveolar fibrin depositions also are frequent findings, according to Idell and associates.64 Coagulation is promoted by the upregulation of endothelial and monocyte and macrophage tissue factor. In a study by Johnson and colleagues,67 the infusion of thrombin in animals caused increased microvascular permeability and lung injury, suggesting that coagulation is involved in the pathogenesis of lung injury rather than being a consequence of such injury. Saldeen101 reported that the inhibition of fibrinolysis contributes to the hypercoagulation seen in ARDS. According to the investigations of Bertozzi and coworkers,12 the inhibition of intra-alveolar fibrinolytic activity is caused in part by the increased release of plasminogen-activator inhibitor type 1.
with ARDS. Intravascular and intra-alveolar fibrin depositions also are frequent findings, according to Idell and associates.64 Coagulation is promoted by the upregulation of endothelial and monocyte and macrophage tissue factor. In a study by Johnson and colleagues,67 the infusion of thrombin in animals caused increased microvascular permeability and lung injury, suggesting that coagulation is involved in the pathogenesis of lung injury rather than being a consequence of such injury. Saldeen101 reported that the inhibition of fibrinolysis contributes to the hypercoagulation seen in ARDS. According to the investigations of Bertozzi and coworkers,12 the inhibition of intra-alveolar fibrinolytic activity is caused in part by the increased release of plasminogen-activator inhibitor type 1.
Alveolar Epithelial Injury
The epithelium of the lung is among the body’s most important tissues, since it occupies 99% of the gas-exchange surface of the lung. One epithelial space is depicted in Figure 75-1. The primary function of the alveolar epithelial or type I cell is to serve not only as a barrier to the outside world but also as a sieve to allow oxygen exchange through the basement membrane. These cells are metabolically active, participating in host defense, alveolar remodeling, and antioxidant functions.
There is a dynamic balance between fluid formation and clearance across the alveolar epithelium, with alveolar liquid volume regulated by a balance between physical and biochemical forces. Factor and associates30 found that ionic movement is dominated by Na+ and Cl–, each moving through specific apical channels. Sodium moves selectively through the epithelial sodium channel (ENaC) and chloride through a channel called the cystic fibrosis transmembrane regulator (CFTR), with adenosine playing a central role by stimulating CFTR-mediated chloride transport.
The clearance of edema fluid from the lung occurs when sodium undergoes centripetal transport from the alveoli into the underlying interstitium. According to Matthay and coworkers,75 this transport appears to be primarily a function of alveolar type II cells. Transport occurs when sodium enters the cell through the epithelial amiloride-sensitive Na+ channel and is pumped out of the cell by Na+, K+-ATPases located on the basolateral surface of the epithelial cells. This enzymatic process of sodium transport may be altered. Folkesson and Matthay34 showed that the Na+ channel inhibitor amiloride will block fluid clearance, whereas the ATPase can be stimulated by beta agonists, increasing sodium transport and therefore transepithelial fluid movement. Chloride is secreted to preserve electrical neutrality and likely moves through the chloride channel.
However, this is not well characterized. Water follows the osmotic gradient generated by Na+ transport. The specific pathway for water movement across the epithelium is controversial. Borok and Verkman14 speculated that it may travel through specialized water-transporting proteins called aquaporins. However, in transgenic mice, where aquaporins have been deleted, Berthiaume and Matthay11 found alveolar liquid clearance to be unaffected.
However, this is not well characterized. Water follows the osmotic gradient generated by Na+ transport. The specific pathway for water movement across the epithelium is controversial. Borok and Verkman14 speculated that it may travel through specialized water-transporting proteins called aquaporins. However, in transgenic mice, where aquaporins have been deleted, Berthiaume and Matthay11 found alveolar liquid clearance to be unaffected.
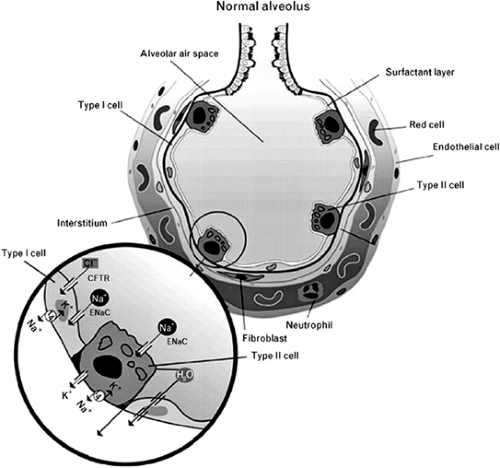 Figure 75-1. Depiction of idealized alveolus, with inset of type I cell or alveolar epithelial cell showing sodium and potassium transport. CFTR, cystic fibrosis transmembrane regulator; EnaC, epithelial sodium channel; Na, sodium; K, potassium; H2O, water. (From Gropper MA, Wiener-Kronish J. The epithelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 2008;14:11.) |
Although alveolar epithelial injury is evident in histopathologic and ultrastructural studies of patients with ARDS, it was not until 1995 that McElroy and associates,77 in an animal model of pneumonia, identified an alveolar epithelial type I cell–specific protein usable as a marker of epithelial cell injury. Goodman and colleagues42 noted that epithelial cell–derived neutrophil activator,78 a second injury marker, is elevated in the bronchoalveolar fluid of patients with ARDS.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


