Acute Myocardial Infarction
Andrew J. Boyle, MBBS, PhD
 ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
![]() Chest discomfort, usually described as “pressure,” “dull,” “squeezing,” or “aching.”
Chest discomfort, usually described as “pressure,” “dull,” “squeezing,” or “aching.”
![]() Characteristic electrocardiographic changes.
Characteristic electrocardiographic changes.
![]() Elevated biomarkers, such as troponin.
Elevated biomarkers, such as troponin.
![]() Imaging may show new regional wall motion abnormality with preserved wall thickness.
Imaging may show new regional wall motion abnormality with preserved wall thickness.
![]() The elderly, women, and diabetics may have atypical presentation.
The elderly, women, and diabetics may have atypical presentation.
 General Considerations
General Considerations
Acute myocardial infarction (MI) is a clinical syndrome that results from occlusion of a coronary artery, with resultant death of cardiac myocytes in the region supplied by that artery. Depending on the distribution of the affected coronary artery, acute MI can produce a wide range of clinical sequelae, varying from a small, clinically silent region of necrosis to a large overwhelming area of infarcted tissue resulting in cardiogenic shock and death. About 1.2 million people experience MI in the United States each year; every minute, one American will die of coronary artery disease.
The risk of having an acute MI increases with age, male gender, smoking, dyslipidemia, diabetes, hypertension, abdominal obesity, a lack of physical activity, low daily fruit and vegetable consumption, alcohol overconsumption, and psychosocial index. As much as 90% of the risk of acute MI has been attributed to the modifiable risk factors. The diagnostic criteria for acute MI are listed in Table 8–1.
Table 8–1. ESC/ACC Definition of Myocardial Infarction

 Pathophysiology & Etiology
Pathophysiology & Etiology
A prolonged imbalance between myocardial oxygen supply and demand leads to the death of myocardial tissue. Coronary atherosclerosis is an essential part of the process in most patients. Ischemic heart disease seems to progress through stages of fatty-streak deposition in coronary arteries to development of fibro-fatty plaque, which then increases in size until it causes luminal obstruction, leading to exertional angina (see Chapter 6). However, at any stage in this process, the atherosclerotic lesion may erode, ulcerate, fissure, or rupture, thereby exposing subendothelial vessel wall substances to the circulating blood. Procoagulant factors (such as tissue factor) reside within the plaque itself and, in the absence of counterbalancing antithrombotic factors (eg, heparin, tissue factor inhibitor) and fibrinolytic activities (tissue plasminogen activator [t-PA] and single-chain urokinase-type plasminogen activator) within the endothelial cells of the coronary artery, can cause thrombosis. This potent procoagulant stimulus results in thrombus development in this region. In general, larger transmural acute MI occurs when this thrombosis propagates and occludes flow within the artery, resulting in ischemia and necrosis of cardiomyocytes distal to the obstruction. This is often associated with ST segment elevation on the electrocardiogram (ECG). Smaller nontransmural MI may occur when platelet thrombus on a plaque embolizes downstream and occludes very small branches. There is usually no ST elevation on the ECG in this situation. Recent work suggests that inflammation may play a pivotal role in the genesis of plaque rupture. Total thrombotic occlusion occurs most commonly in proximal coronary arteries; its presence has been documented during the first 4 hours after infarction in more than 85% of patients with ST segment elevation.
A similar type of myocardial insult occurs occasionally despite angiographically normal coronary arteries and may be caused by emboli (eg, in patients with prosthetic valves or those with endocarditis), dissection of the coronary artery (most commonly in pregnant women), or coronary vaso-spasm (on rare occasions). It can also be caused by thrombosis in situ, the probable mechanism by which patients who have variant angina or who abuse cocaine can suffer acute infarction. In these cases, vasoconstriction secondary to endothelial dysfunction and a propensity to thrombosis are of sufficient magnitude and duration to cause thrombus formation. Oxygen consumption and possibly direct myocyte toxicity also increase with cocaine use. In addition, thrombosis in situ can apparently cause infarction among women who take estrogens (especially if they smoke). An increasingly recognized differential diagnosis of acute MI is stress cardiomyopathy (also known as apical ballooning syndrome or takotsubo cardiomyopathy). This entity can present with a variety of symptoms and ECG changes, including ST elevation, and there is akinesis of the anterior and inferior walls and apex of the left ventricle in the absence of coronary artery disease. It is often accompanied by severe emotional stress. The diagnosis is one of exclusion, after angiography demonstrates patent coronary arteries. The treatment is supportive and the prognosis is good: recovery of ventricular function is the norm.
In addition to blockage of coronary arteries (reduced “supply”), acute MI may be seen when myocardial oxygen requirements are elevated (increased “demand”). This often occurs when other medical illnesses coexist with ischemic heart disease. Pulmonary embolism, pneumonia, arrhythmia, septic shock, severe anemia, or great emotional distress can increase myocardial oxygen demand, reduce coronary perfusion pressure, or evoke paradoxical coronary artery responses and lead to MI. However, these tend to be smaller infarctions with no ECG ST elevation that are diagnosed by elevated biomarkers.
Libby P, et al. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–43. [PMID: 11877368]
Roger VL, et al. Heart Disease and stroke statistics—2012 update. A report from the American Heart Association. Circulation. 2012;125:e12–e230. [PMID: 22179539]
Wittstein IS, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352(6):539–48. [PMID: 15703419]
 Clinical Findings
Clinical Findings
A. Symptoms & Signs
Chest discomfort is the most common symptom; it is usually described as “pressure,” “dull,” “squeezing,” or “aching,” although it may be described differently because of individual variability, differences in articulation or verbal abilities, or concomitant disease processes. The discomfort is usually in the center of the chest and commonly radiates to the left arm or the neck. However, it may also radiate to the right arm, epigastrium, jaw, teeth, or back. The nature of the pain may lead patients to place a hand or fist over the sternum (Levine sign). These clinical signs and symptoms were originally defined in groups of males. It is now clear that women often have more atypical symptoms.
Associated symptoms may include dyspnea, nausea (particularly in inferior infarction), palpitations, and a sense of impending doom.
Patients, especially those with diabetes or hypertension, may have atypical presentations; for example, a diabetic person may have abdominal pain that mimics the discomfort commonly associated with gallstones. In elderly patients, heart failure is often the presenting symptom. Up to 42% of women and 30% of men present with MI without chest pain. The diagnosis of MI should be considered in patients in whom symptoms are atypical yet compatible with ischemia (paroxysms of dyspnea, for example) or in those with atypical chest discomfort. Patients can also have discomfort that is sharper or that radiates to the back. These patients can have pericarditis alone, pericarditis induced by infarction, or a dissecting aortic aneurysm—with or without concomitant infarction.
B. Physical Examination
The physical examination is a critical and underappreciated part of the initial assessment of patients with suspected acute MI. Findings may vary tremendously, from markedly abnormal, with signs of severe congestive heart failure (CHF), to totally normal.
On general inspection, most patients with a large MI appear pale or sweaty and may be agitated or restless. Heart rate should be measured for arrhythmia, heart block, or sinus tachycardia. This is crucial before administration of β-blockers. Assessment of blood pressure is important because severe hypertension (which may be due to the pain) is a contraindication to fibrinolytic treatment and must be treated emergently. Conversely, hypotension in the setting of acute MI may be due to cardiogenic shock, which alters treatment strategy. Fibrinolytic treatments are not effective in cardiogenic shock, and the patient should be considered for an urgent intra-aortic balloon pump (IABP) and primary percutaneous coronary intervention (PCI).
The jugular venous pulse should be carefully examined. Its elevation in the setting of inferior MI without left heart failure suggests right ventricular MI. Detection of right ventricular MI is vital because it portends a much worse prognosis than isolated inferior MI, and the management strategy is different than isolated inferior MI. Whereas elevated jugular venous pulse in left ventricular MI with left ventricular failure may respond to diuresis, right ventricular MI may require intravenous fluid therapy to maintain left ventricular filling.
Cardiac auscultation should be specifically targeted to complications of MI (see later discussion in this chapter) and detection of important comorbidities. Acute MI may result in ischemic mitral regurgitation with a soft S1 and a pansystolic murmur. Acquired ventricular septal defect (VSD) may also result in a pansystolic murmur, but it is usually loud and high-pitched and has a normal S1 and usually occurs later (see later Complications of Myocardial Infarction section). Both ischemic mitral regurgitation and acquired VSD may result in heart failure. The presence of a pericardial friction rub may indicate established infarction that has happened days earlier. Heart failure due to large infarctions may result in a third heart sound. Signs of left heart failure, such as rales and pulmonary hypertension, should also be sought. Important comorbidities, such as concomitant severe aortic stenosis, should also be documented because they may change the initial reperfusion strategy from fibrinolysis or PCI to cardiac surgery with coronary artery bypass graft (CABG) and aortic valve replacement simultaneously.
Alternative diagnoses may also be suggested by clinical examination. The presence of atrial fibrillation or prosthetic valve may suggest that thromboembolism to the coronary artery is the cause of the coronary occlusion. Furthermore, a brief assessment of pulse equality and blood pressure in both arms should be performed. Inequalities in perfusion between arms may indicate aortic dissection, causing compromised blood flow in the branch vessels of the aortic arch. Aortic dissection may also be responsible for occlusion of the ostium of the coronary artery causing the acute MI. This is a surgical emergency and should not be treated with anticoagulation or fibrinolysis. Therefore, a focused clinical examination is an essential part of the initial patient assessment and can be invaluable in guiding therapy.
C. Diagnostic Studies
1. Electrocardiography—The most rapid and helpful test in assessing patients with suspected acute MI is the 12-lead ECG. It should be performed as soon as possible, preferably within 10 minutes, after the patient’s arrival in the emergency department or clinician’s office, since the presence or absence of ST elevation determines the preferred management strategy. For a diagnosis of ST elevation MI (STEMI), ST elevation must be present in at least two contiguous leads. For anterior MI, the precordial (V) leads demonstrate ST elevation (Figure 8–1), and if there is lateral wall involvement, I and aVL may also show ST elevation. In inferior MI, leads II, III, and aVF are affected.
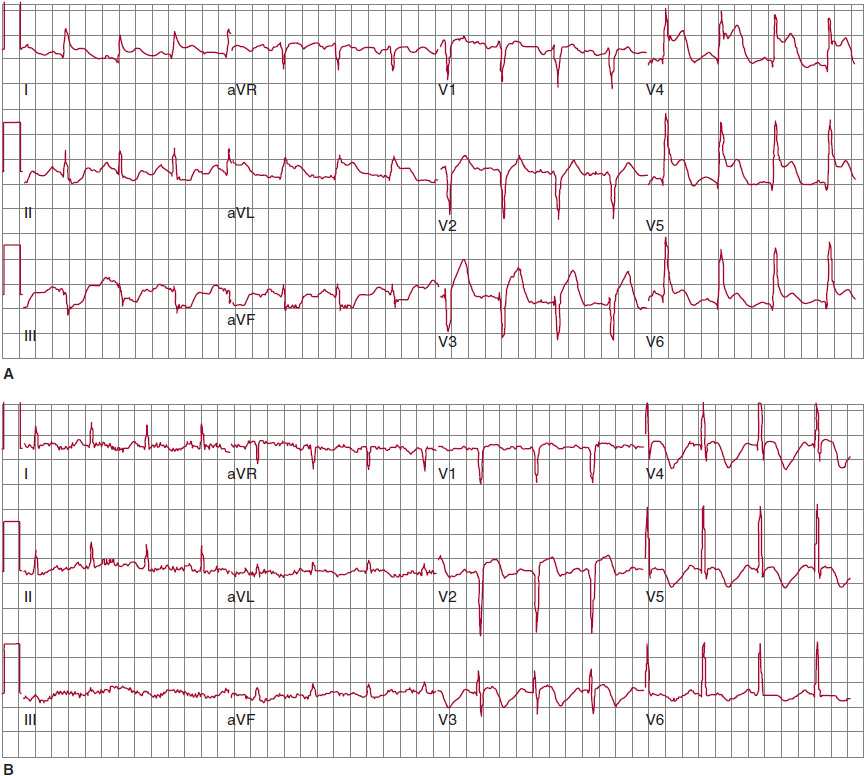
![]() Figure 8–1. Electrocardiogram (ECG) changes in anterolateral ST elevation myocardial infarction (STEMI). A: Initial ECG on presentation shows ST segment elevation in the precordial leads, as well as I and aVL, indicative of acute anterolateral STEMI due to proximal left anterior descending (LAD) coronary artery occlusion. Note the reciprocal ST depression in the inferior leads. B: Following reperfusion, subsequent ECG 48 hours later demonstrates resolution of both the anterolateral ST elevation and the reciprocal changes. Note the Q wave in V2 and the development of T-wave inversion.
Figure 8–1. Electrocardiogram (ECG) changes in anterolateral ST elevation myocardial infarction (STEMI). A: Initial ECG on presentation shows ST segment elevation in the precordial leads, as well as I and aVL, indicative of acute anterolateral STEMI due to proximal left anterior descending (LAD) coronary artery occlusion. Note the reciprocal ST depression in the inferior leads. B: Following reperfusion, subsequent ECG 48 hours later demonstrates resolution of both the anterolateral ST elevation and the reciprocal changes. Note the Q wave in V2 and the development of T-wave inversion.
In addition to standard ECG leads, right ventricular leads should be recorded in all patients with inferior MI. Inferior MI is usually caused by occlusion of the right coronary artery, which may also cause right ventricular infarction. Differentiating right ventricular infarction from left ventricular infarction is imperative because the management is different.
In posterior MI, usually due to circumflex artery occlusion, the only changes seen on a standard ECG may be reciprocal ST depression and R waves (reciprocal of Q waves) in the anterior leads. This ECG pattern should prompt the use of posterior ECG leads V7–9, which may show ST elevation. Even in the absence of ST elevation in posterior leads, true posterior infarction pattern on ECG in the presence of symptoms suggestive of MI should be treated like STEMI.
Non-ST elevation MI (NSTEMI) has a variable presentation on ECG. There may be no ECG changes, or patients may have ST depression, T-wave flattening, or T-wave inversion. Preexisting abnormalities like T-wave inversion may also “pseudo-normalize” in NSTEMI, making it even more difficult to diagnose on ECG. Because determining whether ECG changes are new or old may be difficult, serial ECGs are necessary to diagnose dynamic changes. In patients with symptoms suggestive of MI and no evidence of ST elevation on ECG, the diagnosis of acute coronary syndrome is made. This encompasses unstable angina pectoris and NSTEMI. The distinction between these two entities is made on the presence or absence of elevated biomarkers of MI.
2. Cardiac biomarkers—The diagnosis of infarction requires increases in molecular markers of myocardial injury (Figure 8–2). Myoglobin release from injured myocardium occurs quite early and is very sensitive for detecting infarction. Unfortunately, it is not very specific because minor skeletal muscle trauma also releases myoglobin. Myoglobin is cleared renally, so even minor decreases in glomerular filtration rate lead to elevation. The other early markers advocated by some are isoforms of creatine kinase (CK). This marker has comparable early sensitivity and specificity to myoglobin. The marker of choice in past years was the MB isoenzyme of creatine kinase (CK-MB). A typical rising-and-falling pattern of CK and CK-MB (in the proper clinical setting) was sufficient for the diagnosis of acute infarction. In the typical pattern of CK release after infarction, the enzyme marker level exceeds the upper bound of the reference range within 6–12 hours after the onset of infarction. Peak levels occur by 18–24 hours and generally return to baseline within no more than 48 hours. However, elevations can occur due to release of the enzyme from skeletal muscle. The lack of a rising-and-falling pattern should raise the suspicion that the release is from skeletal muscle, which is usually due to a chronic skeletal muscle myopathy. Elevations of CK in patients with hypothyroidism (where clearance of CK is slowed) and those with renal failure (where clearance is normal because CK is not cleared renally) are caused, in part, by myopathy.
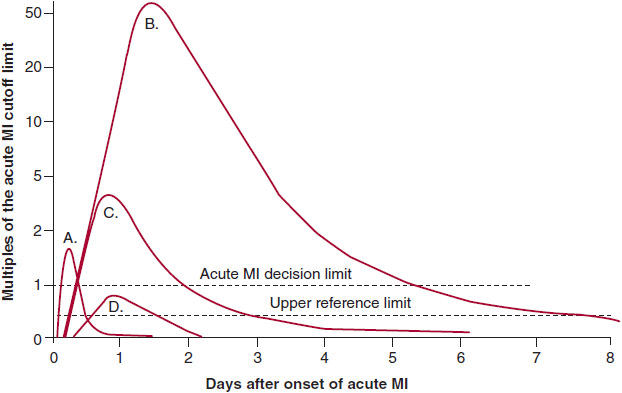
![]() Figure 8–2. Biomarkers in acute myocardial infarction (MI). Plot of the appearance of cardiac markers in blood versus time after onset of symptoms. Peak A, early release of myoglobin or CK-MB isoforms after acute MI. Peak B, cardiac troponin after acute MI. Peak C, CK-MB after acute MI. Peak D, cardiac troponin after unstable angina. Data are plotted on a relative scale, where 1.0 is set at the acute MI cutoff concentration. (Reprinted, with permission, from Wu AH, et al. Clin Chem. 1999;45:1104.)
Figure 8–2. Biomarkers in acute myocardial infarction (MI). Plot of the appearance of cardiac markers in blood versus time after onset of symptoms. Peak A, early release of myoglobin or CK-MB isoforms after acute MI. Peak B, cardiac troponin after acute MI. Peak C, CK-MB after acute MI. Peak D, cardiac troponin after unstable angina. Data are plotted on a relative scale, where 1.0 is set at the acute MI cutoff concentration. (Reprinted, with permission, from Wu AH, et al. Clin Chem. 1999;45:1104.)
Cardiac troponins I and T are proteins found in cardiac muscle cells and released into the circulation from damaged cardiac myocytes during acute MI. Troponin levels (either I or T) are significantly more sensitive and specific for myocardial damage than CK. Troponin becomes detectable in serum between 4 hours and 6 hours after onset of an acute MI, peaks and then falls to lower levels, and remains elevated at these low levels for 5–7 days (see Figure 8–2). Thus, the late or retrospective diagnosis of acute MI can be made with this marker, making the use of lactate dehydrogenase isoenzymes superfluous. Therefore, because of its sensitivity and specificity for cardiac muscle damage as well as its early rise and continued low-level detectability, troponin is the preferred biomarker for diagnosis of acute MI. Furthermore, it has been shown to correlate with prognosis even in the absence of CK or CK-MB elevation. Newer high-sensitivity troponin markers are in development, and these have the potential to detect myocardial necrosis at lower levels and more rapidly.
Coronary recanalization, whether spontaneous or induced pharmacologically or mechanically, alters the timing of all markers’ appearance in the circulation. Because it increases the rapidity with which the marker is washed out from the heart, leading to rapid increases in plasma, the diagnosis of infarction can be made much earlier—generally within 2 hours of coronary recanalization. Although patency can be approximated from the marker rise, distinguishing between thrombolysis in myocardial infarction (TIMI) II and TIMI III flow is not highly accurate. It should also be understood that peak elevations are accentuated, which must be taken into account if the clinician wants to use peak values as a surrogate for infarct size.
3. Imaging—In the emergency setting, most diagnoses of acute MI are made on history, physical examination, and ECG. However, when the history is atypical and the ECG is equivocal or uninterpretable, performance of a rapid bedside echocardiogram may demonstrate a new regional wall motion abnormality with preserved wall thickness, suggestive of acute MI. In most cases of STEMI, however, echocardiography is not warranted because it delays reperfusion therapy. Echocardiography is also helpful in diagnosing complications of MI, such as VSD, papillary muscle rupture or free wall rupture, and tamponade.
In NSTEMI that is diagnosed on elevated plasma levels of cardiac biomarkers, nuclear scintigraphy or echocardiography may help determine the region of the heart affected by the MI, but these are not standard diagnostic tools.
Canto JG, et al. Association of age and sex with myocardial infarction symptom presentation and in-hospital mortality. JAMA. 2012;307:813–22. [PMID: 22357832]
Hassan AKM, et al. Usefulness of peak troponin-T to predict infarct size and long-term outcome in patients with first acute myocardial infarction after primary percutaneous coronary intervention. Am J Cardiol. 2009;103:779–84. [PMID: 19268731]
Menown IB, et al. Early diagnosis of right ventricular or posterior infarction associated with inferior wall left ventricular acute myocardial infarction. Am J Cardiol. 2000;85(8):934–8. [PMID: 10760329]
Thygesen K, et al. Third universal definition of myocardial infarction. J Am Coll Cardiol. 2012;60(16):1581–98. [PMID: 22958960]
 Treatment
Treatment
The goals of treatment in acute MI are stabilization of the patient and salvage of as much myocardium as possible. A number of general measures should be performed in all patients. In patients with ST elevation—who are at highest risk for complications and have ongoing cardiomyocyte necrosis—immediate reperfusion of the infarct artery should be attempted. The management of acute MI is summarized in Table 8–2.
Table 8–2. Overview of Management of Acute Myocardial Infarction

Early recognition of symptoms of myocardial ischemia may lead to faster treatment and salvage of myocardium. Therefore, it is recommended that patients at risk for MI be educated about the symptoms suggestive of acute MI and call for emergency help immediately if they have these symptoms.
A. Prehospital Management
Aspirin, 162–325 mg, should be given immediately. Continuous cardiac monitoring, oxygen, and sublingual nitroglycerin should be administered to all patients with suspected acute MI. Communities usually have organized protocols for ambulance personnel regarding (1) whether or not they should obtain a 12-lead ECG, (2) whether there are designated hospitals that receive patients in whom an acute MI is suspected, or (3) whether the patient should be taken to the nearest emergency department. In some regions of the world, fibrinolytic treatment is initiated in the ambulance, based on a 12-lead ECG.
B. Emergency Department Therapy
On arrival, all patients with suspected acute MI should have a 12-lead ECG performed immediately. If aspirin has not been given, then 162–325 mg of aspirin should be administered immediately. All patients with suspected MI should have continuous cardiac ECG monitoring, and intravenous access (two separate intravenous lines) should be gained in all patients. Sublingual nitroglycerin and intravenous morphine should be administered if patients have active chest pain. Oxygen saturations should be monitored noninvasively, rather than by arterial blood gas measurement. Supplemental oxygen, 2–4 L/min, should be given to all patients, particularly if they are hypoxemic. A portable chest radiograph should be ordered but should not delay reperfusion, unless a diagnosis of aortic dissection is strongly considered. Echocardiography may be considered if the diagnosis of MI remains in doubt (eg, equivocal history, uninterpretable ECG). Oral β-blockers should be administered to all patients with acute MI, unless there is a contraindication, such as hypotension, bradycardia, or asthma. This has been shown to improve outcomes and limit the size of infarction. Intravenous β-blockers could be considered when there is hypertension or tachyarrhythmia, for example (Table 8–3). However, they should be avoided in patients with signs of heart failure, in those with contraindications to β-blockers, and in those at high risk for cardiogenic shock (age < 70 years, heart rate > 110/min or < 60/min, systolic blood pressure < 120 mm Hg, or prolonged time since the onset of symptoms).
Table 8–3. Standard Doses of Commonly Used Agents in Patients with Acute Myocardial Infarction
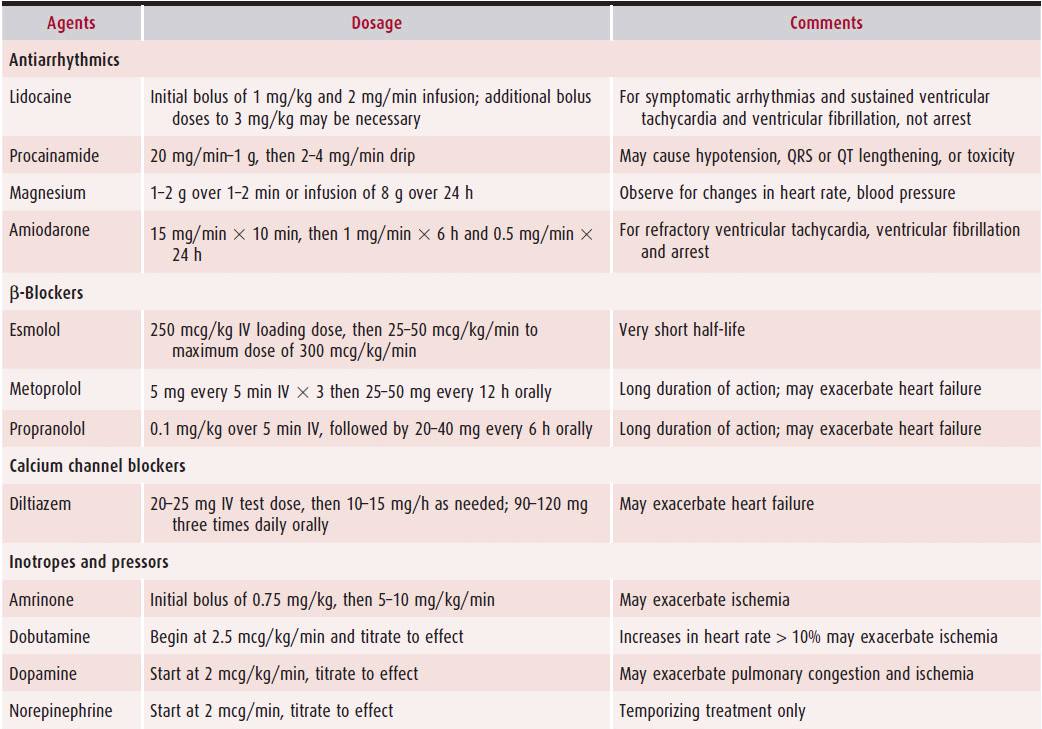
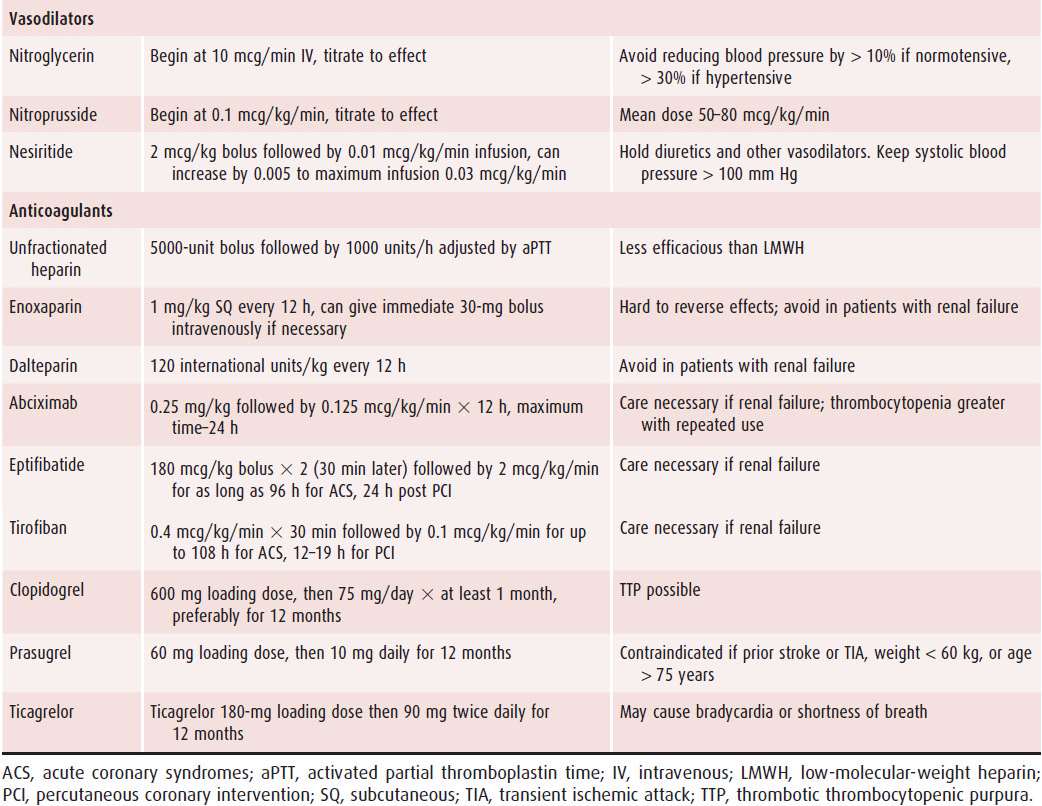
An anticoagulant should be administered to all patients with acute MI, unless a contraindication exists. In patients with ST elevation who receive fibrinolytics, 48 hours of anticoagulant should be administered. Acceptable choices for anticoagulants include unfractionated heparin (UFH), low-molecular-weight heparin (LMWH), and fondaparinux. For patients with ST elevation undergoing primary PCI, acceptable choices for anticoagulants include UFH, LMWH, and bivalirudin. UFH is preferred to LMWH in most institutions for primary PCI because it has a short half-life, it can be turned off rapidly if there is a complication during invasive therapy, and it can be monitored during procedures with a bedside activated clotting time test. In contrast, LMWHs have a long half-life, and there is no bedside test of their anticoagulant efficiency.
C. Reperfusion Therapy
1. Patients with STEMI—All patients with STEMI who seek medical care within the first 12 hours after symptom onset should be considered for urgent reperfusion of the infarct-related artery, but the earlier therapy is begun, the greater the benefit. In addition, those patients who seek medical care within 12–24 hours of symptom onset may be considered for reperfusion, particularly if chest pain is ongoing or heart failure or shock has developed, but the benefit of reperfusion therapies after more than 12 hours is less well established. The definitive therapies for reperfusion in STEMI are fibrinolysis or PCI. Both of these strategies improve patency of the infarct-related artery, reduce infarct size, and lower mortality rates. Therefore, one or the other method should be performed as quickly as possible. The goal of reperfusion therapies in the United States is a door-to-needle time of 30 minutes (for fibrinolysis) and a door-to-balloon inflation time of less than 90 minutes (for PCI). PCI has been shown to be superior to fibrinolysis when it is performed without significant delay by experienced clinicians in experienced centers (Figure 8–3). However, significant delays in performing PCI reduce its benefit over fibrinolytic therapy. There are special cases where primary PCI is always preferred over fibrinolysis: cardiogenic shock, severe CHF or pulmonary edema (Killip class III), or if there are contraindications to fibrinolysis (Table 8–4). These patients may require insertion of an IABP and may benefit from mechanical reperfusion with primary PCI. The different management of patients with these high-risk clinical features underscores the need for careful clinical examination of all patients with chest pain.
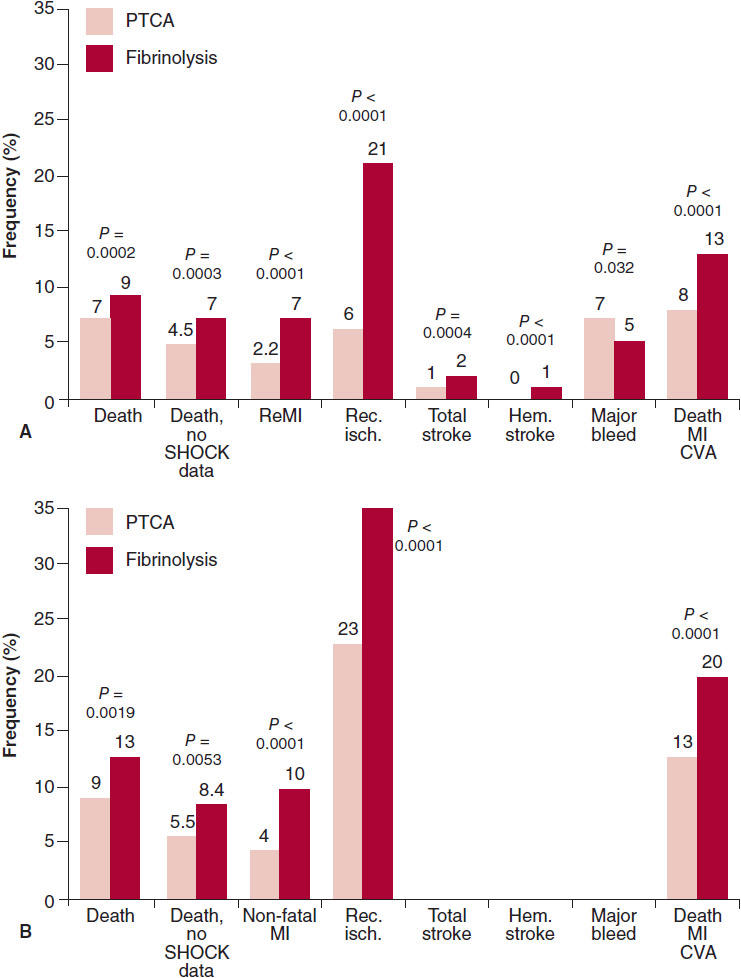
![]() Figure 8–3. Percutaneous coronary intervention (PCI) versus fibrinolysis for ST elevation myocardial infarction (STEMI). Short-term (4–6 weeks; A) and long-term (B) outcomes for various end points shown are plotted for STEMI patients randomized to PCI or fibrinolysis for reperfusion in 23 trials (n = 7739). Primary angioplasty for acute STEMI improves both short- and long-term outcomes. CVA, cerebrovascular accident; Hem. stroke, hemorrhagic stroke; Rec. isch., recurrent ischemia; ReMI, recurrent myocardial infarction. (Modified, with permission, from Keeley EC, et al. The Lancet. 2003;361:13 and Antman EM, et al. J Am Coll Cardiol. 2004;44:671.)
Figure 8–3. Percutaneous coronary intervention (PCI) versus fibrinolysis for ST elevation myocardial infarction (STEMI). Short-term (4–6 weeks; A) and long-term (B) outcomes for various end points shown are plotted for STEMI patients randomized to PCI or fibrinolysis for reperfusion in 23 trials (n = 7739). Primary angioplasty for acute STEMI improves both short- and long-term outcomes. CVA, cerebrovascular accident; Hem. stroke, hemorrhagic stroke; Rec. isch., recurrent ischemia; ReMI, recurrent myocardial infarction. (Modified, with permission, from Keeley EC, et al. The Lancet. 2003;361:13 and Antman EM, et al. J Am Coll Cardiol. 2004;44:671.)
Table 8–4. Contraindications for Fibrinolysis Use in STEMI

Recent data suggest that all patients with STEMI, whether they undergo primary PCI or fibrinolytic therapy, benefit from early administration of a thienopyridine in addition to aspirin. Acceptable alternatives include clopidogrel, prasugrel (only if undergoing primary PCI), or ticagrelor. However, thienopyridines may cause an increase in bleeding complications if the patient undergoes CABG surgery.
2. Patients with NSTEMI—These patients should not be treated with fibrinolytics. The definitive management of NSTEMI involves anticoagulation, platelet inhibition, and consideration for an early invasive strategy (ie, routine coronary angiography with or without PCI during the index hospitalization).
For detailed discussion of NSTEMI, see Chapter 7.
D. In-Hospital Management
All patients with acute MI should be admitted for continuous cardiac monitoring. Patients should have bed rest for the first 12–24 hours following MI and reperfusion, but in the absence of ongoing ischemia, they should be mobilized after this time. All patients should receive the appropriate cardiac diet, adhering to the National Cholesterol Education Program (NCEP) Adult Treatment Panel III (ATP III) dietary guidelines, as well as education regarding the necessary dietary changes they should make after discharge.
Within the first 24 hours of presentation, the long-term medical management of patients with both STEMI and NSTEMI should be commenced. Angiotensin-converting enzyme (ACE) inhibitors should be given on day 1, if the patient’s blood pressure allows, particularly in those with anterior MI or impaired left ventricular function. ACE inhibitors reduce left ventricular remodeling and heart failure and should be continued long-term. β-Blockade should have already been started in the emergency department and should be continued orally in all patients, unless there are absolute contraindications, and should also be continued long-term. Aspirin, 162–325 mg daily, should be administered initially, then 81 mg daily for life. Thienopyridines (clopidogrel, prasugrel, or ticagrelor) should be continued for 12 months. Hydroxymethylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (statin therapy) should be given soon after MI and should be continued at high dose long-term.
During hospitalization, all patients should be educated about adhering to therapeutic lifestyle changes, including dietary and lifestyle measures, smoking cessation, and medication compliance. Patients should be referred to a cardiac rehabilitation program to consolidate these messages and develop an appropriate exercise regimen. This has been shown to reduce mortality after MI.
E. Primary PCI Versus Fibrinolysis
The goal of reperfusion is to rapidly restore blood flow to the myocardium to prevent ongoing ischemic cell death. Therefore, whichever means can achieve reperfusion most quickly should be used. Primary PCI results in improved patency rates of the infarct-related artery, as well as improved TIMI flow grade, compared with fibrinolysis. In general, the patency rate with primary PCI is 90% or higher, whereas with thrombolysis, the rate is about 65% and recurrent events are more common. With modern advances, coronary stenting has further improved long-term outcomes over balloon angioplasty alone. PCI has therefore been widely accepted as the treatment of choice for STEMI in centers that can perform primary PCI rapidly and effectively (Figure 8–4). However, very early after the onset of symptoms, when the thrombus in the infarct-related artery is still soft, fibrinolysis may recanalize the artery as quickly as, if not more so than, primary PCI. This is true in the first hour and possibly the first 3 hours after symptom onset. Therefore, fibrinolysis is an acceptable treatment in these early time points. However, after 3 hours, primary PCI has a clear benefit over fibrinolysis and should be considered the preferred therapy. It bears restating that primary PCI should only be performed in centers skilled in the treatment of STEMI that can achieve rapid reperfusion, with a goal door-to-balloon inflation time of 90 minutes.
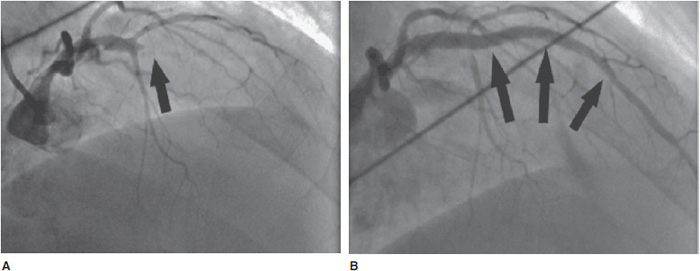
![]() Figure 8–4. Primary percutaneous coronary intervention (PCI) for acute myocardial infarction (MI). A: Initial angiography of a patient presenting with acute anterior ST elevation myocardial infarction (STEMI) shows an occluded left anterior descending (LAD) coronary artery (arrow). B: Following angioplasty and stenting, patency of the LAD is restored.
Figure 8–4. Primary percutaneous coronary intervention (PCI) for acute myocardial infarction (MI). A: Initial angiography of a patient presenting with acute anterior ST elevation myocardial infarction (STEMI) shows an occluded left anterior descending (LAD) coronary artery (arrow). B: Following angioplasty and stenting, patency of the LAD is restored.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


