Acute and Chronic Responses to Toxic Inhalations
INTRODUCTION
The lungs and airways are in constant contact with the outside world and thus, are especially vulnerable to toxic substances present in the environment. Within seconds of exposure to an inhaled toxin, pathologic events occur that may cause immediate distress, systemic illness lasting days, or even lead to the development of chronic lung disease. This chapter discusses the pathology and pathophysiology that can result from various inhaled toxins, and also highlights the role of several common and medically significant toxic inhalants that are known to cause acute and chronic pathophysiologic responses in the lung. The chapter also discusses several systemic syndromes caused by acute toxic inhalations. The scope of this chapter does not include chronic exposure to low levels of toxins.
DETERMINANTS AND MECHANISMS OF IRRITANT-INDUCED PULMONARY INJURY
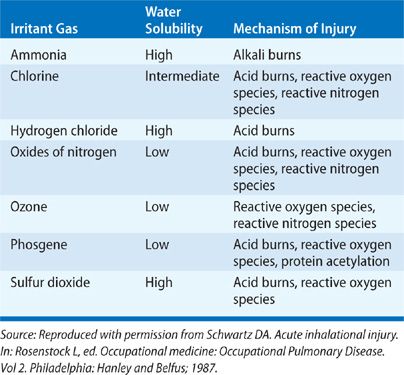
Inhaled toxins exist in many forms and may be categorized by taking into account their physical properties. General categories include gases, vapors, fumes, aerosols, and smoke. A variety of factors determine the pathologic results of a toxic inhalation: the size of inhaled particles, the solubility of the inhaled substance in water, the concentration of the inhalant in ambient air, the duration of exposure, the presence or absence of ventilation, and a variety of host factors (age, smoking status, comorbid diseases, use of respiratory protection, and perhaps even genetic susceptibility). While toxic inhalants provoke a broad range of chemical and biologic activities that contribute to pathogenesis, their physical properties, namely their particle size and water solubility, are of fundamental importance in determining the site and severity of pulmonary injury. Tables 90-1 to 90-3 summarizes the physical properties of the discussed inhalants that substantially affect the resulting pathogenesis of these agents.
Source: Adapted with permission from Kizer KW. Toxic inhalations. Emerg Med Clin North Am. 1984;2(3):649–666.
The size of aerosolized particles is of critical importance in inhaled toxin pathogenesis. In general, larger aerosolized particles are more likely to deposit on the nasopharynx via impaction and not gain access to the lower airways, while smaller particles are able to penetrate smaller airways and effect toxicity at the level of the alveolus. Aerosolized particles larger than 30 to 80 μm are not inhalable through the nose,1 and particles larger than 5 μm typically do not reach the alveoli.2 Ultrafine particles (those <0.1 μm) have been specifically implicated in the toxicity due to the agents of polymer fume fever.3 Inhaled particles may have direct toxic effects themselves, or they may function as vehicles for adsorbed gaseous agents that are toxic to terminal bronchioles and alveolar cells.
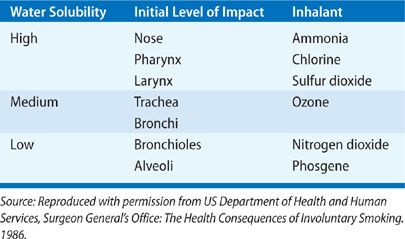
In addition to particle size, the relative solubility of an inhalant in water determines where along the respiratory tract toxicity will occur. Substances with high water solubility, such as ammonia, sulfur dioxide, and hydrochloric acid, provoke immediate and evident injury to the conjunctiva and mucosal surfaces of the upper airways; they are largely absorbed by the mucus lining the pharynx and larynx and often react there to form caustic acids and alkalis. The provoked symptoms quickly prompt exposed individuals to flee the area or contain the source of exposure, reducing the duration of exposure. These compounds can also activate irritant receptors in the upper airways, provoking a bronchoconstrictor reflex that may further limit access of the inhalant to lower airways.4 In contrast, compounds like phosgene and ozone have low water solubility and thus fail to cause immediate irritation, promoting longer exposure to the inhalant and deeper penetration of the lower airways. Compounds of intermediate solubility, for example, chlorine gas, typically have pathologic effects throughout the respiratory tract. These differences in solubility can be overcome by differences in concentration and duration of inhalant exposure: virtually any inhaled toxin (even the most soluble agents) can cause diffuse damage of the respiratory tract by overwhelming the absorptive capacity of the upper respiratory tract. Furthermore, adsorption of a toxic gas on particulate matter may permit a toxin access to otherwise unreachable airways.
Host factors also play a significant role in predicting an individual’s response to a toxic inhalation. Underlying pulmonary or extrapulmonary disease may worsen a patient’s response to an exposure. Children deposit a smaller fraction of inhaled particles in their nasopharynx than adults and thus may be at elevated risk of lower airway exposure and pathology5; moreover, as some gases (such as chlorine and sulfur dioxide) are heavier than air, children may be subjected to a longer duration and higher concentration of gas than adults near the same site of toxin release. With particles greater than 0.5 μm, breathing through one’s nose increases upper airway particle deposition compared to mouth-breathing; this difference is absent with particles less than 0.5 μm.6 Tobacco smoking impairs ciliary clearance and cellular defense, limiting the exposed patient’s ability to clear inhaled particles and prolonging exposure. Patients with increased minute ventilation (such as those panicking at the scene of an irritant gas release) are at elevated risk of increased exposure and toxicity. An emerging literature in experiments with inbred strains of mice suggests that genetic variants may alter the risk of responding to various inhaled toxins.7
Injury from toxic inhalation may occur via a number of mechanisms. If the concentration of the inhalant is high enough and if ventilation is inadequate, simple asphyxiation due to displacement of atmospheric oxygen may occur. The reflex bronchoconstriction triggered by upper airway irritant receptor activation may itself cause inadequate oxygen inhalation. Cell injury from acute toxin exposure typically occurs via nonimmunologic mechanisms of injury and inflammation, generally via formation of an acid (chlorine, oxides of nitrogen, phosgene, and sulfur dioxide), an alkali (ammonia), or reactive oxygen or nitrogen species (ozone, oxides of nitrogen and chlorine). Acid formation results in coagulation of underlying tissue, while alkali exposure causes a liquefaction of mucosa and characteristically deep lesions within the airways. Reactive oxygen and nitrogen species and their derivatives achieve local tissue damage via lipid peroxidation and protein oxidation, and may cause similar toxicity systemically.8 Free radicals may be direct derivatives of inhaled substances, or they may be released by alveolar macrophages that are activated by inhalant exposure.9 All three types of tissue damage generally lead to an increase in expression of proinflammatory cytokines that can perpetuate the acute injury and may be responsible for the development of later sequelae. Disruption and repair of injured airway epithelial tissue may compromise the host’s defenses against further infectious or irritant substances. A role played by the innate immune system in disease progression is evident in the case of endotoxin exposure in organic dust toxin syndrome (ODTS, below), and may be a host factor in the response to ozone and nitrogen dioxide.7,10
PATHOGENESIS AND CLINICAL PRESENTATION OF TOXIC INHALATION INJURY
Mechanisms of disease and clinical manifestations based on respiratory site exposure are considered below.
 UPPER AIRWAY
UPPER AIRWAY
Effects of toxins on the upper airways are typically sudden and short-lived compared to those more distal along the respiratory tract; chronic pathology in this region is unusual. Compounds that provoke a response in the nose, pharynx, and larynx tend to be particulate with relatively large average particle size or gases with high water solubility. Acids, alkalis, and reactive oxygen and nitrogen species may all cause tissue injury in this region depending on the inhaled compound and its reactions along airway epithelium. Characteristic tissue injury depends on dosage and ranges from slight edema of the nasopharynx and larynx to epithelial ulceration and frank hemorrhage. Once the airway epithelia are compromised, it fails to function as a protective barrier against the environment. Underlying inflammatory cells, nerves, muscles, and blood vessels become exposed, which may further the inflammatory response. An obstructive response to some irritants starts to occur at concentrations only barely perceivable as irritating.11
The typical presentation of patients with acute exposure of irritant substances to the upper airways includes burning sensations of the nasal passages and throat, copious sputum production, coughing, and sneezing. Extrapulmonary manifestations include burning of the eyes, profuse lacrimation, headache, and dizziness. The most serious risk in the exposed patient is airway obstruction due to reflex bronchospasm or laryngospasm, mucosal edema, increased secretions, and sloughed epithelial cells. Patients presenting with hoarseness or stridor should be carefully observed for further evidence of airway compromise. Though inhalational injury confined to the upper airways tends to be self-limited with no or few long-term sequelae, a chronic rhinitis following irritant exposure, reactive upper airway dysfunction syndrome (RUDS) has been described12 and was observed among World Trade Center rescue workers following the attacks of September 11, 2001.13
Patients with acute toxic exposure to the upper airways should be immediately removed from the source, which may require removal of the patient’s clothes. The patient’s airway should be secured and monitored; racemic epinephrine may be used, but it should not delay endotracheal intubation if necessary. Frequent suction may be required. Profuse amounts of water should be irrigated over exposed surfaces. Supplemental oxygen should be provided if appropriate. Patients with extensive upper airway edema may benefit from corticosteroids,14 though this is unsupported by clinical trials. Ophthalmologic consultation should be sought for management of eye exposure.
 CONDUCTING AIRWAYS
CONDUCTING AIRWAYS
Acute or chronic effects on the airways may be observed.
Acute Injury
As is the case with the upper airways, the conducting airways protect their submucosal structures with epithelium that may be compromised by acute inhalational injury. The resulting edema, inflammation, and bronchoconstriction may be life-threatening if it results in an obstructed airway, and without its epithelial barrier the airway is vulnerable to infections and other environmental pathologies. This damage to the epithelium appears to occur at the tight junction interface between cells,15–17 resulting in increased epithelial permeability to other irritants, which gain direct access to effector cells within the subepithelial mucosa. Resulting bronchospasm may cause ventilation–perfusion mismatch. The smooth muscle of the airways can be hyperresponsive in the hours and days following irritant exposure, an effect probably mediated by the neutrophilic and eosinophilic inflammatory response inhalational injury provokes.18,19
Conducting airway injury may manifest as intrathoracic airflow obstruction hours after the initial insult. Patients with histories of exposure who present with any evidence of respiratory compromise should be hospitalized for observation, even if asymptomatic. Findings of concern include expiratory wheezing, decreased airflow on peak expiratory flow measurement or spirometry, abnormalities of gas exchange or an abnormal chest X-ray. Likewise, patients with complaints of dyspnea or chest tightness should be observed carefully and treated symptomatically with inhaled steroids and bronchodilators, even in the absence of objective findings. When significant airflow obstruction is present, systemic steroids may be of some utility.
Chronic Injury
Two forms of chronic injury include reactive airways dysfunction syndrome and vocal cord dysfunction.
Reactive Airway Dysfunction Syndrome A persistent asthma-like disease following acute exposure to an irritant inhalant, known as reactive airway dysfunction syndrome (RADS, or “Brooks syndrome”) was named in 198520 but observed among World War I soldiers exposed to war gases.21 Investigation and diagnosis of the disease is generally limited by the absence of spirometry results in patients prior to exposure and the presence of confounding factors (e.g., cigarette smoking), but numerous reports exist of previously asymptomatic patients experiencing hyperreactive airway disease presenting soon after a single toxic exposure and persisting for months or years.20,22–29 RADS is distinguished from immunologic occupational asthma in that it follows a single exposure and does not follow a latency period of sensitization to the offending substance.
The pathogenesis of RADS likely begins with the initial injury to and desquamation of the epithelium, which results in hemorrhage and edema followed by inflammatory changes, and finally long-term structural changes of the airways involving epithelial regeneration and fibrosis.30,31 Ensuing airway narrowing may be due to mucosal edema, inflammation, or structural changes to the architecture of the bronchial wall.32
RADS typically presents abruptly within 24 hours following exposure with the classic symptoms of obstructive airway disease: wheezing, chest tightness, dyspnea, and cough. The symptoms and obstructive findings on examination and spirometry are relieved by bronchodilators, though not as effectively as in other types of reactive airway disease (perhaps due to chronic fibrotic remodeling of the conducting airways). The disease can persist for months and, may, in some instances, be permanent.33 A recent case report shows that a patient with severe unremitting symptoms of RADS for 5 years following an ammonia exposure responded to high-dose oral vitamin D supplements.34 Anecdotally, inhaled corticosteroids have shown benefit in relieving airflow obstruction.30 Systemic corticosteroids have been proven beneficial in an animal model of RADS.35
Vocal Cord Dysfunction Vocal cord dysfunction may also follow a single acute irritant exposure and may be confused with RADS.36,37 The disorder may be caused by reflex response to nerve stimulation by irritants.38 Patients suspected of having RADS who do not respond appropriately to bronchodilators should be evaluated for vocal cord dysfunction; direct laryngoscopy is the gold standard for diagnosis.37
 LOWER AIRWAYS AND PULMONARY PARENCHYMA
LOWER AIRWAYS AND PULMONARY PARENCHYMA
The lower airways and lung parenchyma are primary sites for inhalational injury. Effects may be acute or chronic.
Acute Injury
Though all toxic inhalants are capable of producing distal airway disease at extreme concentrations and durations, the gases most likely to do so are those with low water solubility like phosgene and nitrogen dioxide, which bypass reflex bronchoconstriction and absorption by upper airway mucus (Fig. 90-1). The initial pathologic events in distal airways are caused by the cellular toxicity of the inhaled agent and its derivatives, which compromise the impermeability of the alveolar–capillary interface.39 Some of this cytotoxicity may be derived indirectly from reactive oxygen species released from activated inflammatory cells.9 In the absence of an intact alveolar–capillary interface, profound pulmonary edema may develop that impairs gas exchange and can prove fatal. The severity of this pulmonary edema, which typically presents after a latent period of several hours following the initial insult, is likely dose-related. This process may cause no more than slight dyspnea and cough with a mild alveolar infiltrate, or it may progress via diffuse alveolar damage to acute respiratory distress syndrome (ARDS). For this reason, patients with exposure to gases capable of causing distal airway disease should be hospitalized and monitored for symptoms of respiratory distress and with serial chest X-rays for at least 24 hours following exposure. Development of ARDS from toxin exposure likely shares a common pathway with other causes of acute lung injury, and management is similar: supportive care with mechanical ventilation, careful control of blood glucose, surveillance for infection, and deep vein thrombosis prophylaxis. Diuresis, intravenous corticosteroids, prone positioning, nitric oxide inhalation, and exogenous surfactant are all unsupported by clinical trials but potentially of some benefit. Diffuse bronchiolitis has also been reported following acute exposure.40
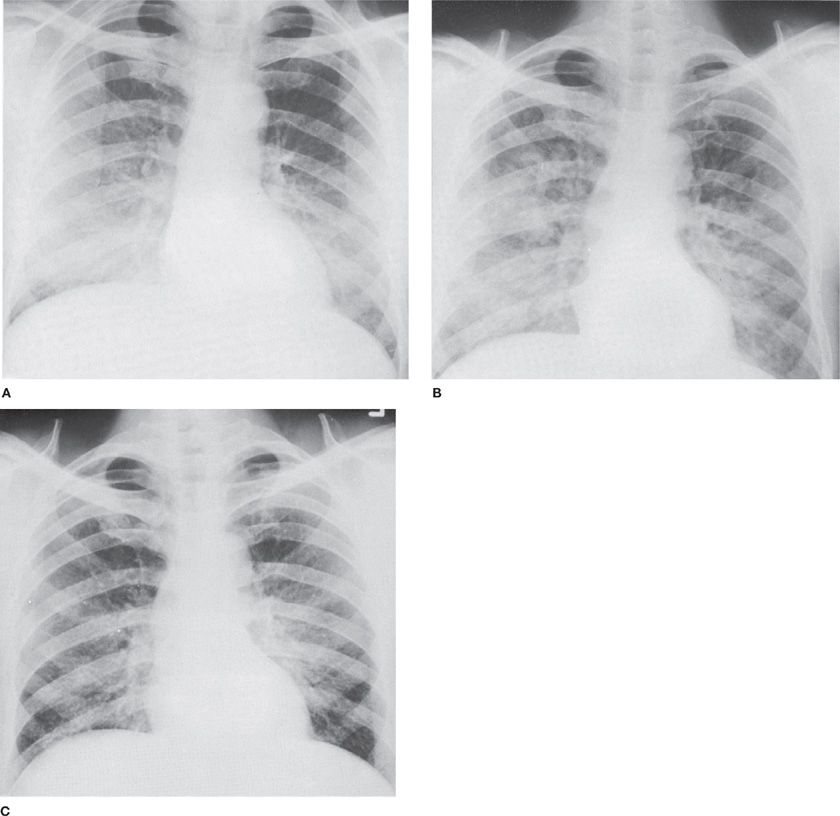
Figure 90-1 Accidental exposure of a 55-year-old mechanic to spill of liquid Cl2, followed immediately by coughing and dyspnea. A. Day of exposure. Bilateral alveolar infiltrates, most marked on right. B. Two days later. Progression of alveolar infiltrates. C. Seven days later. Incomplete resolution of infiltrates associated with persistent shortness of breath.
Chronic Injury
Two forms of chronic injury include bronchiolitis obliterans and bronchiolitis obliterans organizing pneumonia.
Bronchiolitis Obliterans Bronchiolitis obliterans (BO) is a well-documented but infrequent long-term sequela of toxic gas exposure, especially of nitrogen dioxide (Refer to Chapter 54)41,42 but also to ammonia,43 mercury,44 and sulfur dioxide.45,46 The disease typically presents 1 to 3 weeks following the initial lung injury and pulmonary edema (Fig. 90-2). The interim is often free of symptoms. When BO does develop, patients may present with dyspnea on exertion or obstructive findings on spirometry. Physical examination may be either unremarkable or remarkable only for early inspiratory crackles. Chest X-ray will either be normal or demonstrate hyperinflation. Pulmonary function tests typically demonstrate airflow obstruction that may in some cases also be associated with restrictive defects. On biopsy, granulation tissue will be seen in the lumen of small airways and bronchiole walls may be obliterated by fibrous scarring. Corticosteroids may be of benefit in preventing or alleviating BO if administered early in the course of the disease,47 though this is controversial.
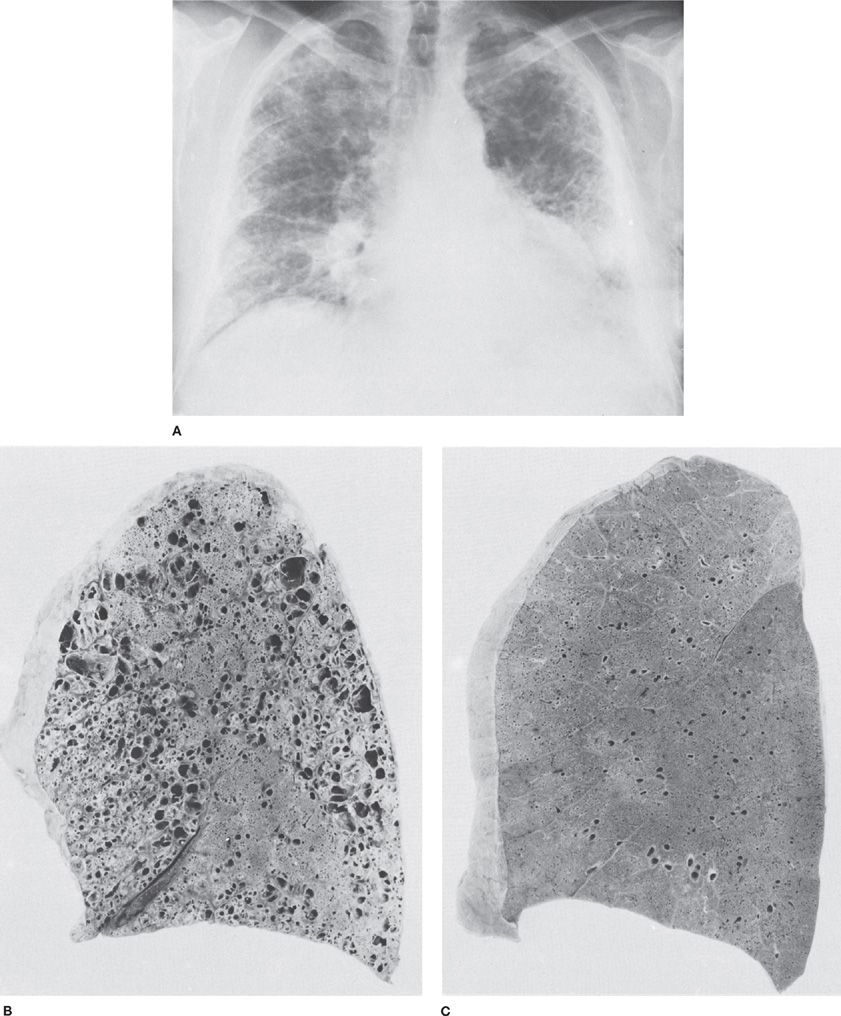
Figure 90-2 Bronchiolitis obliterans in a 63-year-old man who had been exposed to a wide variety of unidentified fumes in his jobs, which included welding. A. Chest radiograph. Diffuse pulmonary fibrosis and honeycombing, most marked in the peripheral portions of the lungs. B. Sagittal section of lung from same patient showing markedly dilated airspaces. Microscopic sections revealed bronchiolitis obliterans and chronic interstitial pulmonary fibrosis. C. Normal lung from a 43-year-old man who died suddenly. The difference between (B) and (C) in the alveolar portions of the lungs is striking. (Used with permission of Dr. R. Ochs.)
BO Organizing Pneumonia BO organizing pneumonia (BOOP) is another observed delayed sequela of toxic inhalation.48,49 Patients present in the weeks following exposure with fever, a persistent and nonproductive cough, sore throat, and malaise. Late inspiratory crackles may be observed. Chest X-ray may reveal bilateral patchy “ground-glass” densities that start as focal lesions but may coalesce with time. Pulmonary function tests generally reveal a restrictive process with decreased diffusion capacity. Histologically, granulation tissue extends past the terminal bronchioles and into the alveolar spaces, sometimes with interstitial scarring. BOOP and BO are probably both chronic results of the initial inflammatory response to the toxic insult and the ensuing proliferative process. BOOP responds well to corticosteroids,50 though a small number of patients may develop progressive fibrosis. Duration of therapy should be guided by the patient’s clinical status.
EFFECTS OF SPECIFIC INHALED TOXINS ON THE RESPIRATORY SYSTEM
The respiratory consequences of inhalation of commonly encountered toxins are discussed individually below.
 AMMONIA
AMMONIA
Ammonia (NH3) is a stable colorless gas at room temperature and a water-soluble nitrogen-containing compound that ranks among the most commonly spilled hazardous substances (Table 90-4B.51 Familiar to all as a household cleaner, it also has countless uses in industry: as a chemical coolant used for refrigeration, as a fertilizer, as a fixative in photocopiers, and in the manufacture of polymers and explosives. Small amounts are naturally present in the atmosphere as products of the putrefaction of vegetable and animal proteins. The smell of concentrated ammonia (as low as 5 ppm) is immediately recognizable. Due to the prevalence of ammonia in household cleaners and to its use in smelling salts (which exploit the noxious effect of ammonia on nasal membranes to arouse consciousness), the odor threshold may extend to over 50 ppm because of the olfactory fatigue and adaption.52,53 Ammonia is frequently dissolved into water for storage and transportation, and vaporizes readily on exposure to air. Most inhalation exposures are the result of accidental releases, including tank leaks and transportation mishaps,54–56 and most exposures occur in the industrial workplace.57 A recently reported source of exposure is via fumes produced in clandestine methamphetamine laboratories.58
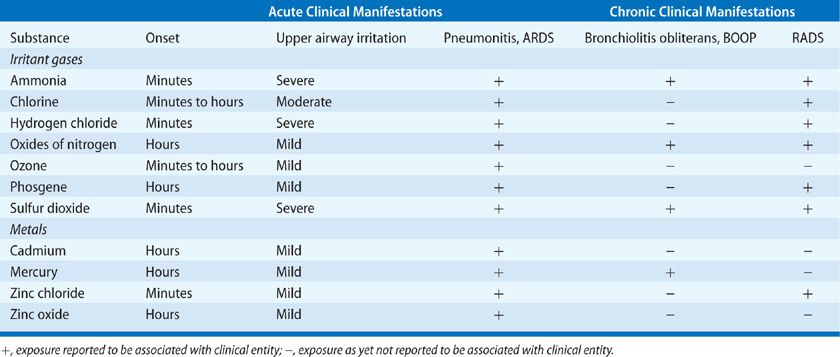
The mechanisms of toxicity to the respiratory system involve both direct irritation and alkaline chemical burns. Ammonia tends to affect the proximal airways, where it reacts rapidly with the water present on mucosal surfaces to form ammonium hydroxide, causing tissue liquefaction. This necrosis liberates formerly intracellular water, which serves as further reactant for ammonia, perpetuating the reaction. In addition to the alkali burns caused by the generated ammonium hydroxide, thermal burns can result from the heat generated by this exothermic reaction. The resulting injury, typical of alkali burns, penetrates deeply. The initial injury to the mucosa of the oropharynx can cause edema, hemorrhage, sloughing of tissue, and increased secretions that can bring about fatal upper airway obstruction. Ammonia is directly caustic to airways at concentrations of 1000 ppm and higher.
Though concentrated at the proximal airways, the effects of ammonia have been observed at all levels of the respiratory tract. The penetration of the gas to the smaller airways and alveoli is a function of its concentration and the duration of exposure. Reported acute conditions associated with ammonia exposure include pulmonary edema, laryngitis/tracheobronchitis, bronchiolitis, and bronchopneumonia; reported chronic sequelae include bronchiectasis, bronchospasm/asthma (termed RADS), and chronic obstructive pulmonary disease.59 There are several reports of interstitial lung disease following a single exposure to ammonia.57 A biphasic pattern of pulmonary response to ammonia inhalation has been reported, characterized by initial, acute pneumonitis that may clear over the next 2 to 3 days, followed in some individuals by the gradual development of airway obstruction and respiratory failure.54 There may be a correlation between the development of a bacterial superinfection after exposure with the ensuing emergence of bronchiectasis.60 In one review of published case reports, 21% of patients with acute ammonia inhalation died within 60 days of exposure.59 The most common causes of death were laryngeal edema and obstruction, noncardiogenic pulmonary edema, and extensive pneumonic complications.
Management of a patient who has experienced ammonia inhalation requires removing him or her from the source of the irritant, securing the airway, and immediately irrigating all exposed surfaces (especially the eyes) with copious amounts of water. Humidified oxygen should be administered early. Airway management aggressiveness depends on the extent of complications such as laryngeal edema, pulmonary edema, hemoptysis, or respiratory failure that occur.61 Rales detected on physical examination are predictive of the subsequent hospital course, even in the absence of hypoxemia and chest X-ray abnormalities.54 Medical management is largely supportive. Aerosolized bronchodilators together with corticosteroids can be administered to treat bronchospasm. Corticosteroids and antibiotics are both frequently used, but both are unproven in human trials.
 CHLORINE, CHLORAMINES, AND HYDROCHLORIC ACID
CHLORINE, CHLORAMINES, AND HYDROCHLORIC ACID
Chlorine (Cl2) is a common gas of intermediate water solubility. The first reports of its toxicity followed its use as an agent of chemical warfare in World War I, and war gassings remain the largest historical source of chlorine gas exposure.62 Most exposures since then have occurred in the industrial setting, where chlorine is used in the manufacture of paper, cloth, antiseptics, and other products.63 More common in the household is the liberation of chloramines and other toxic chlorine derivatives from the reaction of chlorine-containing products (such as hypochlorite bleach) with ammonia or products containing hydrochloric or phosphoric acid. Numerous exposures to chlorine gas have occurred near swimming pools, where chlorine-releasing agents (e.g., calcium hypochlorite and chlorinated isocyanuritic acids) are used in water purification.40 Chlorine gas is greenish-yellow in color and is heavier than air; though its odor is distinct, patient exposure to it may be prolonged compared to other toxic gases due to its delayed irritation of mucosal surfaces and its high density, which keeps it low to the ground.
The pathogenicity of chlorine gas derives directly from elemental chlorine’s effects on the respiratory tract and indirectly from its reaction with water to form hydrochloric acid (HCl) and hypochlorous acid (HOCl). The character and distribution of injury from chlorine exposure varies according to duration of exposure and the relative concentrations of elemental chlorine and its derivative compounds. HCl and HOCl possess considerable water solubility and are responsible for the tissue damage sustained by the upper airways and ocular conjunctivae. Irritation to trigeminal nerve endings caused by these compounds can cause a reflex bronchoconstriction that may contribute to compromise airway diameter.64 In addition to causing the tissue coagulation typical of acid exposures (described above), these compounds ionize and enter cells, where they may form reactive oxygen species. HOCl has been shown to react with nitrite (NO2–) to produce reactive nitrogen-containing compounds able to nitrate, chlorinate, and dimerize phenolic amino acids.65 As nitrite and nitric oxide (its parent compound) levels are elevated at sites of tissue inflammation, this potentially is another mechanism of injury.66 Though lower respiratory tract irritation has been reported following high-level exposures, less than 5% of inhaled chlorine gas penetrates beyond the upper airways.67 Fatal dosages from chlorine inhalation have ranged from 50 to 2000 ppm.40
The immediate clinical manifestations of acute chlorine exposure are typical of irritants of its solubility: rhinitis, cough, dyspnea, wheezing, and chest tightness, along with conjunctivitis and skin irritation.68 When chlorine gas exposure has resulted acutely in death, autopsies have revealed diffuse ulcerative tracheobronchitis, pulmonary edema, thrombi within pulmonary vessels, and denudation of respiratory tract epithelium.69 Acute respiratory symptoms are more prevalent and severe among patients who already have chronic respiratory disease. In 2005, a derailment train released 40 to 60 tons of chlorine gas near a small town in South Carolina. Nine people died and 71 were hospitalized. The majority of people (>90%) had respiratory symptoms including wheezing, rales/crackles, cough, decreased breathing sound, rhonchi, and labored breathing. Autopsy findings revealed the causes of death included asphyxia, lactic acidosis, and acute respiratory failure.70
The lasting respiratory sequelae of chlorine gas exposure have been described since the years following use of the gas in World War I.71 Reported long-term pulmonary diseases following exposure have included both restrictive and obstructive processes, frequently resolving to normal function within a month and almost always before 2 years following exposure. RADS may be an infrequent sequela of high-level exposures to chlorine.22,72
Patients who have been exposed to chlorine gas should be managed according to the severity of their presenting symptoms similarly to other victims of irritant inhalation. The treatment for lung injury is primarily supportive care including humidified oxygen administration, lung protective mechanical ventilation, and pharmacologic therapy. Nebulized sodium bicarbonate has shown promise as a useful treatment,73 but lacks supporting clinical trials and showed no outcome benefit in a relatively large observational study of chloramine gas exposure.74 Beta-agonist bronchodilators and corticosteroid are frequently used and are probably of benefit. The reported benefit of corticosteroid administration is anecdotal69 and unconfirmed by clinical trial.
 SULFUR DIOXIDE
SULFUR DIOXIDE
Sulfur dioxide is a heavy, colorless, and highly water-soluble gas that has the distinct, pungent odor of burnt matches. It is generated in the combustion of coal and petroleum and is often used as a preservative in alcoholic beverages and fruit. Industrial exposures have occurred around ore smelting, metal smelting, oil refining, sugar refining, and the bleaching of wool and wood pulp. In 1930, 63 people died and thousands became ill in the narrow Meuse River Valley of Belgium due to the trapped high concentrations of sulfur dioxide from the waste products of industry resources (United States Environmental Protection Agency). Sulfur dioxide is among the most harmful gases released to the atmosphere during volcanic eruptions75; in 1986 the gases emitted from one eruption killed nearly 2000 people in Cameroon.76 Sulfur dioxide’s great density keeps it low to the ground and slow to dissipate from sites of release; thus children may be at an increased risk of exposure.
Sulfur dioxide reacts with water present on mucus membrane to form sulfuric acid, which causes tissue coagulation underlying exposed surfaces. Sulfuric acid also further dissociates into hydrogen ions, sulfite, and bisulfite,77 which can then react with oxygen to produce reactive oxygen species; ensuing lipid peroxidation may be a contributing mechanism of injury to immediate tissues and elsewhere.8,78
Sulfur dioxide is detectible to humans at 3 to 5 ppm and is lethal at levels exceeding 400 ppm for 1 minute.79 High water solubility makes sulfur dioxide primarily an upper airway irritant. The direct irritant of bisulfate iron inhibits mucociliary transport.80 The irritation-induced parasympathetic stimulation leads to smooth muscle contraction and mucosal secretion. Exposures of high enough intensity can irritate both upper and lower airways. Patients exposed typically present with dyspnea, burning of the nose and throat, rhinorrhea, cough, and airway obstruction.81 Proximal airway injury is characterized by acute denudation of the airway mucosa without inflammatory cell infiltrates. When lower airways are exposed, alveoli fill with fluid due to noncardiogenic pulmonary edema and the clinical picture is consistent with ARDS. Alveolar architecture is generally preserved. Extremely high-intensity acute exposures can lead within minutes to death from respiratory failure due to a combination of alveolar hemorrhage and edema, possible reflex vagal stimulation, and the asphyxiating effect of high concentrations of sulfur dioxide.82 RADS has been reported following single sulfur dioxide exposure29; bronchitis has also been observed.83 One pattern of postexposure progression reported is a rapid recovery followed several weeks later by the onset of irreversible airflow obstruction due to BO.82 Care for patients who have been exposed to sulfur dioxide is supportive: humidified supplemental oxygen, bronchodilators, and intubation and ventilation if necessary. The use of corticosteroids in the setting of ARDS following sulfur dioxide exposure has not been shown to be of benefit, but a trial is not unreasonable. Antibiotics may be reserved for use upon evidence of infectious complications.
 NITROGEN OXIDES
NITROGEN OXIDES
Nitrogen oxides are ubiquitous air pollutants, released from automobile engines and the combustion of coal and petroleum, and are present in cigarette smoke. High-level acute exposure is most likely to occur in industrial settings, including mining, acetylene welding, and explosives manufacturing. A well-known form of exposure occurs in “Silo-filler’s disease,” in which farmers inhale concentrated nitrogen dioxide gas released within silos by decomposing nitrogenous biomaterial.41,84 Exposures to high levels of nitrogen dioxide have been attributed to blast furnaces,40 anesthetic gases,85 military incidents,86,87 and ice-hockey arenas (secondary to combustion byproducts from ice-resurfacing machines).88–91 Nitrogen dioxide is a liquid at room temperature and a reddish-brown gas above 70ºF. The acrid smell of nitrogen dioxide makes the inhalation exposure to be avoided at low concentration. Thus, short-term (1 hour peak) nitrogen dioxide concentration outdoor is unlikely to exceed 0.2 ppm.92 However, low concentrations at 4 ppm may anesthetize the nose, resulting in overexposure.
Nitrogen dioxide is hydrolyzed by the water on mucosal surfaces to form nitric and nitrous acids, though much of its toxicity is explained via the free radical activity of nitrogen dioxide itself and the nitrites and nitrates that derive from it.93 Though the predominant site of toxicity from nitrogen dioxide exposure is the interface of the terminal bronchioles and alveolar membranes,94 the relative insolubility of nitrogen dioxide in water ensures that enough gas penetrates the upper airways such that injury can occur virtually anywhere along the respiratory tract.95 Nitrogen dioxide itself is a reactive nitrogen species that, along with other reactive derivatives, is capable of lipid peroxidation and protein oxidation,96 both of which may be significant contributors to the gas toxicity via disruption of the cell membrane. Mice with defective toll-like receptor 4 expression exhibit a lessened response to nitrogen dioxide exposure compared to normal strains,10 suggesting that the patient’s innate immunity may play a role in disease development. There is also evidence that nitrogen dioxide exposure is mutagenic to lung cells.97
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


