Acid–Base Balance
Regulation of [H+] is of crucial importance for maintenance of normal cellular functions. The normal [H+] is maintained at about 40 nEq/L. When there is even a small change in the [H+], intracellular proteins gain or lose H+ ions resulting in alterations in charge distribution which may affect molecular structure and protein function. The hydrogen ion concentration in bodily fluids is largely regulated by the ratio of the concentrations of carbon dioxide and bicarbonate. This is predicated upon the relationship demonstrated in the Henderson–Hasselbalch equation:
![]()
where pH = –log[H+] (the H+ concentration measured in moles per liter) and pKa = 6.10. The lungs are responsible for modulating arterial PCO2, whereas the kidneys are primarily responsible for modulating the concentration of bicarbonate in plasma. In concert, these organs maintain a stable extracellular acid–base milieu that is readily assessed by measuring arterial pH.
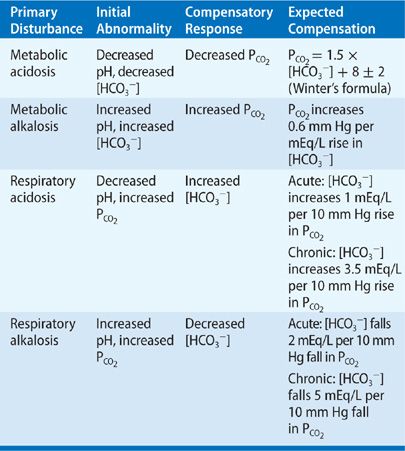
The normal internal environment is maintained within narrow limits: The arterial blood pH is kept remarkably close to 7.40, the bicarbonate concentration is maintained around 24.5 mEq/L, and the PCO2 is maintained at about 40 mm Hg. Deviations of the pH with accompanying changes in the PCO2 and [HCO3–] result in the four major categories denoted in Table 17-1. Metabolic acidosis is characterized by acidemia (pH < 7.35) that is due to reduced plasma [HCO3–]. Metabolic alkalosis is characterized by an alkalemia (pH > 7.45) that results from an elevation in the plasma [HCO3–]. Respiratory acidosis is due to hypoventilation resulting in a net increase in PCO2 (hypercapnia) and a concomitant fall in pH. Respiratory alkalosis is due to primary hyperventilation leading to a fall in PCO2 (hypocapnia) and a rise in pH.
TABLE 17-1 Patterns of PCO2 and HCO3 Changes in Acid–Base Disorders
In this chapter, we first review the basic physiologic roles that the kidneys and lungs play in maintaining acid–base balance and then discuss their adaptation in primary acid–base disorders. The following section then focuses on clinical application of physiologic concepts in analyzing acid–base problems as encountered by the clinician.
BASIC PHYSIOLOGY OF THE ROLE OF THE KIDNEY IN ACID–BASE BALANCE
Normal metabolism generates large quantities of volatile acid (CO2) and nonvolatile acid daily. The complete metabolism of carbohydrates and fats generates 15,000 mmol of CO2 daily. This leads to acid generation as the CO2 combines with H2O to form carbonic acid (H2CO3). As the volatile fraction is excreted by the lungs during respiration, acid accumulation does not occur. The nonvolatile or “fixed” fraction is produced at a rate of 1 mEq/kg per day. The major source of the nonvolatile acid fraction is the oxidation of sulfur-containing proteins from the diet to sulfuric acid. If this amount of nonvolatile acid is not excreted, life-threatening metabolic acidosis ensues; therefore, for a normal individual to maintain acid–base balance, 50 to 100 mEq of nonvolatile acid must be excreted daily by the kidneys.
The addition of 50 to 100 mEq of acid requires initial buffering before it can be excreted. Whole-body buffering capacity is composed of interacting buffer systems: the bicarbonate and nonbicarbonate buffers (Buf–), consisting primarily of hemoglobin, proteins, and phosphates. The sum of the buffer anions [HCO3–] and [Buf–] is the total buffer base and defines total-body buffering capacity. Since all body buffer systems are in equilibrium, a change in the serum [HCO3–] reflects concurrent changes in the other body buffer systems. The importance of bicarbonate in buffering is due to its relationship with CO2. As H+ ions are buffered by HCO3–, there is a decrease in the [HCO3–] and a concurrent increase in the dissolved [CO2]. As the [CO2] can be excreted by the lungs to maintain a constant [CO2], this substantially increases the buffering capacity of bicarbonate. Since the kidney plays a major role in controlling the [HCO3–] and [HCO3–] is easily measured in serum, the HCO3– anion is a useful parameter to evaluate the renal response to an acid load.
The H+ ions released from the dissociation of sulfuric acid are titrated by blood bicarbonate and nonbicarbonate buffers.
![]()
Although the added H+ is excreted via CO2 elimination by the lungs, this occurs at the cost of depletion of [HCO3–]. To replenish the consumed base, bicarbonate is reabsorbed by the kidneys and returned to the blood. This process does not accomplish the replacement of consumed base, since continuous metabolic production of acid will ultimately decrease the available base present. The process of renal regeneration of base requires the urinary excretion of acid or H+ ions in the absence of any urinary bicarbonate. For every H+ ion excreted, a bicarbonate is returned to the body. If there is any bicarbonate in the urine, there will be a net gain of H+. Therefore the kidney has two major functions in this context: (1) reabsorption of all the filtered bicarbonate—this takes place primarily in the proximal tubule and (2) the base consumed by metabolism must be generated in the process of urinary acid excretion. This takes place in the distal portions of the nephron, the distal collecting tubule, and the collecting ducts.
BICARBONATE RECLAMATION
The proximal tubule is responsible for reclaiming 70% to 90% of the filtered bicarbonate. This may occur either by direct bicarbonate absorption at the proximal tubule or via proton secretion into the lumen of the tubule. The latter mechanism appears to be the predominant pathway.1 Acid excretion across the apical membrane of the proximal tubule occurs by an Na+/H+ antiporter (NHE3)2 and to a lesser extent by a proton pump. The secreted proton enters the tubular fluid and combines with filtered bicarbonate ions leading to carbonic acid formation. Under the influence of carbonic anhydrase, carbonic acid is then split into CO2 and H2O. The CO2 diffuses into the cell where it is rehydrated to carbonic acid and then again split into protons and bicarbonate. The bicarbonate ion exits from the cell through the basolateral membrane into the interstitium via a 3HCO3/Na (NBCe1) symporter, while the proton is secreted into the lumen. The basolateral membrane Na+/K+ ATPase antiporter, maintaining a low intracellular sodium concentration, further enhances the NHE3 activity.
In summary, reabsorption of bicarbonate is a cyclic phenomenon and requires carbonic anhydrase and is strictly associated to sodium reabsorption.
Biochemical studies show that total NHE3 and NBCe1 protein abundance are upregulated by chronic respiratory acidosis.3 However, the main mechanism responsible for the elevation in serum bicarbonate is the increased excretion of titratable acid and ammonium which are stimulated by persistently elevated PCO2 (see Fig. 17-1).4 It is important to understand that this process reclaims filtered bicarbonate but does not result in a net gain of bicarbonate. At the end of the proximal tubule there is a lowering of the luminal pH from 7.26 to 6.70, and the bicarbonate concentration is lowered from 24 mEq/L to 8 mEq/L.5 The fluid delivered to the distal tubule is essentially the same with respect to pH and bicarbonate concentration as that which leaves the proximal tubule. The reclamation of the remaining bicarbonate occurs in the thick ascending limb and in the outer medullary collecting tubule. At the collecting tubule, H+ secretion occurs primarily by an H+ ATPase pump at the luminal membrane and bicarbonate entry to the blood is via a Cl–/HCO3– exchanger at the basolateral membrane.6
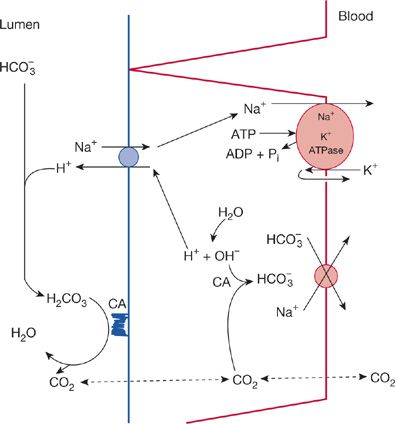
Figure 17-1 Schematic representation of proximal tubular reclamation of filtered bicarbonate. In the lumen, filtered bicarbonate reacts with secreted H+, generating carbonic acid, which is dehydrated by carbonic anhydrase, CA, located on the brush border. The cell secretes H+ by a process that exchanges H+ for filtered Na+. The source of secreted H+ is water, which in turn generates OH– and subsequently bicarbonate because of the presence of intracellular CA. Bicarbonate exits the basolateral side of the cell linked in some fashion with Na+; sodium is also actively pumped out of the cell.
The crucial role of carbonic anhydrase is demonstrated by the fact that carbonic anhydrase inhibitors, that is, acetazolamide, result in bicarbonate wasting and the generation and maintenance of metabolic acidosis. The most physiologically important regulators of reclamation of bicarbonate are the pH, the PCO2, and the extracellular volume status of the patient. In states of acidosis, there is enhanced luminal Na+/H+ exchange that may be mediated by an increase in intracellular H+ ions and by increasing the number of new exchangers and increased activity of the Na+/HCO3– cotransporter at the basolateral membrane. Elevation of the PCO2 will promote higher proximal tubular concentration of CO2 and lead to intracellular acidosis, giving rise to further secretion of H+ ions and reclamation of bicarbonate. If there is volume depletion, there will be avid Na+ reabsorption at the proximal tubule in exchange for H+ and thus greater reabsorption of bicarbonate. Other factors that are important include the luminal bicarbonate concentration, the tubular flow rate, and the serum potassium.
NET RENAL ACID EXCRETION
Net excretion of acid occurs primarily in the distal nephron and is largely mediated by the active secretory pumps, H+-K+ ATPase and H+ ATPase. The latter appears to be linked in some way to Cl– reabsorption to preserve electroneutrality. By definition, to produce net H+ excretion the secreted H+ will have to be excreted in processes that do not consume bicarbonate.
To achieve net secretion of protons in the luminal fluid of the distal nephron requires association of the protons with urinary buffers other than bicarbonate. Although secreted protons lower the urinary pH to 4.5 resulting in a 3 pH unit differential from arterial pH (a 1000-fold increase in H+ concentration), the quantity of acid excreted as free H+ is trivial. For example, daily excretion of 2 L of urine with a pH of 5 would result in excretion of only 0.02 mEq of dissociated H+ ions in contrast to the 50 to 100 mEq of H+ generated each day from dietary sources. The nonbicarbonate buffers present in the urine that carry out the role of net acid excretion are the titratable buffers, primarily phosphate, which accounts for 40% of net acid excretion, and ammonia, which accounts for the remainder.
The ability of phosphate to act as proton acceptor in the urine is based on its pKa of 6.8. As the urine pH is lowered below the pKa of 6.8, there is conversion of HPO4– to H2PO4. This transfer continues until the urine pH reaches 5.5, at which point almost all the phosphate present is in the associated form, H2PO4. Other components of this system are uric acid (pKa = 5.75) and creatinine (pKa = 4.97). Although the titratable buffers account for a sizable fraction of net basal acid excretion, they cannot increase in amount to enhance acid excretion in settings of acid loading since phosphate excretion depends on phosphate intake and not on synthesis as is the case for ammonia excretion.
The rate of ammonium (NH4+) production and excretion can, however, be varied according to physiologic needs. Ammonia (NH3) combines with H+ to form ammonium, which is trapped in the collecting tubule lumen and excreted in the urine. The pKa for this reaction is 9.0. The majority of ammonia is synthesized in the proximal tubular cell by the enzymatic breakdown of glutamine. Glutamine is actively taken up by the proximal tubule at the apical and basolateral membranes and transported to mitochondria.1 Deamidation by glutaminase forms ammonium and glutamate. The latter is further metabolized by glutamate dehydrogenase to form ammonium and α-ketoglutarate. Metabolism of α-ketoglutarate to bicarbonate in the liver leads to return of bicarbonate to the systemic circulation (Fig. 17-2).
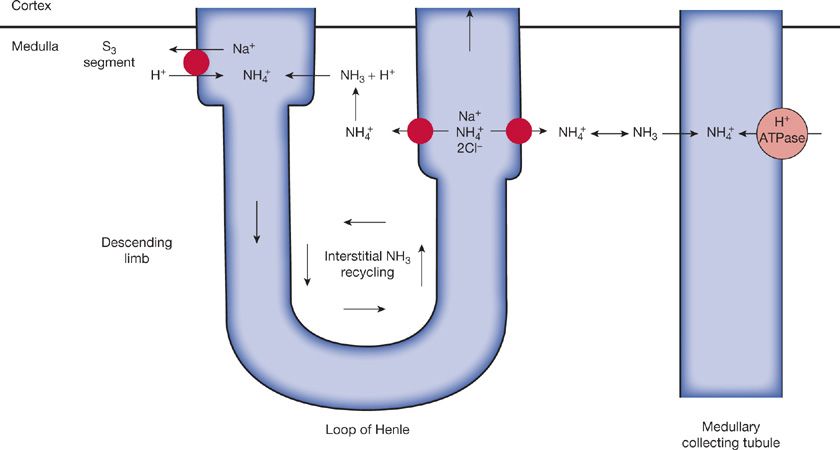
Figure 17-2 Schematic representation of ammonia recycling within the renal medulla. Although NH4+ production occurs predominantly in the proximal tubule, most of the NH4+ is then reabsorbed in the thick ascending limb, apparently by substitution for K+ on the Na+-K+-2Cl– carrier in the luminal membrane. Partial dissociation into NH3 and H+ then occurs in the less acid tubular cell. The NH3 diffuses into the medullary interstitium, where it reaches relatively high concentrations; it then diffuses back into those segments that have the lowest pH and therefore have the most favorable gradient: the S3 segment of the late proximal tubule and, more important, the medullary collecting tubule, where the secreted NH3 is trapped as NH4+ and then excreted. (Reproduced with permission from Rose B. Clinical Physiology of Acid-Base and Electrolyte Disorders, 4th ed. New York: McGraw-Hill; 1994.)
The ammonium that is formed is transported into the proximal tubular lumen via the Na+-H+ antiporter, working in this case as an Na+–NH4+ antiporter. The ammonium is then reabsorbed in the thick ascending limb by substitution of NH4+ for K+ on the Na+–K+–2Cl– carrier. The intracellular ammonium in the thick ascending limb cell is then dissociated into ammonia and H+. The ammonia accumulates in the medullary interstitium and is finally secreted into the lumen of the medullary collecting tubule. At this site, due to the low lumen pH (4.5–5) the ammonia accepts an H+ and is trapped in the lumen and excreted in the urine as NH4Cl.
The importance of the ammonia system is that it can be regulated by the systemic acid–base state. An acid load initially leads to an increase in ammonium excretion within 2 hours due to formation of a more acidic urine which enhances ammonia diffusion into the lumen at the collecting duct. After 5 to 6 days there is maximal NH4+ excretion due to increased glutamine uptake and enhanced activity of phosphate-dependent glutaminase and glutamate dehydrogenase to produce more ammonium in the proximal tubule.6 This is presumably mediated by intracellular acidosis of the proximal tubular cell. The net effect is that NH4+ excretion can increase from about 30 mEq per day to as much as 300 mEq/d in severe metabolic acidosis. The plasma potassium is an important regulator of ammonia synthesis as hyperkalemia will result in a transcellular influx of K+ in exchange for H+ resulting in lowering of the intracellular H+ concentration, thus causing an intracellular alkalosis with consequent inhibition of ammonia synthesis. Hypokalemia would have the opposite effect. Urinary acidification is also very important, since an inability to lower urinary pH will result in a reduction in NH3 trapping in the collecting duct lumen and a subsequent inhibition of the degree of ammonium formation. Inadequate acidification of the urine will also inhibit H2PO4 formation.
RESPIRATORY CONTRIBUTION TO ACID–BASE BALANCE
The major roles of the lungs in acid–base balance are to excrete the CO2 produced daily by aerobic metabolism and to compensate for primary metabolic acid–base disturbances by altering the rate and depth of ventilation. The CO2 generated by the tissues diffuses into the plasma, at the peripheral capillaries, and is present in the blood in three compartments. Part of the CO2 remains in the gas phase, but the amount is limited by the solubility coefficient of CO2 (0.03 mM/mm Hg). CO2 may also react with amino groups of proteins and form carbamino compounds. The majority of the CO2 is carried within red blood cells.7 The red cells contain carbonic anhydrase, which hydrates the CO2 and thus forms carbonic acid, which dissociates to H+ and HCO3–. The protons are buffered by hemoglobin which has an increased affinity for H+ at the low oxygen tension present in the peripheral capillaries and venous blood. The bicarbonate produced in the red cell leaves the cell in exchange for chloride. This chloride shift is a characteristic response to elevation of CO2 in the blood resulting in an acute elevation of bicarbonate in exchange for a drop in serum chloride. When the blood enters the pulmonary circulation, the enhanced oxygenation of hemoglobin promotes release of bound H+. The H+ and HCO3–, via carbonic anhydrase, combine to reform CO2, which passively diffuses from the blood into the pulmonary interstitium where the CO2 tension is very low. Subsequently, CO2 is lost into the alveolar space.
The rate of minute ventilation is controlled by two sets of chemoreceptors: Those in the respiratory center in the brain stem and those in the carotid and aortic bodies located at the bifurcation of the carotid arteries and in the aortic arch, respectively. The central chemoreceptors are stimulated by an increase in the PCO2 or by metabolic acidosis, both of which appear to be sensed by a fall in the pH of the surrounding cerebral interstitial fluid. The peripheral chemoreceptors are primarily stimulated by hypoxemia, although they may also respond to acidemia. The level of alveolar or effective ventilation varies in accord with the total minute ventilation. Level of total ventilation changes as a function of metabolic demand. Under normal circumstances, PCO2 is well controlled between 38 and 42 mm Hg according to the relationship:
![]()
where ![]() is CO2 production (reflecting metabolic rate) and
is CO2 production (reflecting metabolic rate) and ![]() is alveolar ventilation (reflecting CO2 clearance).
is alveolar ventilation (reflecting CO2 clearance).
Under basal conditions the volatile acid production or CO2 that is metabolically generated is completely eliminated by the lungs. The mechanism of the central stimulation of respiration in response to an elevated CO2 is a topic of intense debate and will not be focused upon in this section (see further details in Chapter 11). However, intracranial adjustments to pH have been consistently observed and have interesting parallels to the effects of acidosis on the proximal tubular cell in the kidney. Increased concentrations of CO2 in the cerebrospinal fluid (CSF) result in intracellular acidosis, an increase in CSF bicarbonate concentration, and an equimolar reduction in CSF chloride concentration.7 As brain cells increase their bicarbonate concentration, there is increased buffering, and intracellular brain pH is returned toward normal. The major group of cells within the central nervous system (CNS) responsible for acid–base regulation are the glial cells and the cells of the choroid plexus.7,8 These cells contain carbonic anhydrase9 which converts intracellular CO2H2O to H+ and HCO3–. The H+ is exchanged for Na+ on the blood side, allowing the intracellular pH to increase. The administration of acetazolamide into the cerebral ventricles blocks the expected increase in CSF bicarbonate in response to hypercapnia.8 In addition to changes in bicarbonate concentration in the CSF in response to hypercapnia, there are also changes in the levels of ammonia.10 Brain and CSF ammonia increase in hypercapnia; ammonia acts to enhance H+ buffering thereby preventing a fall in the bicarbonate concentration.
ACUTE AND CHRONIC ADAPTATION TO RESPIRATORY ACIDOSIS
Figure 17-3 depicts the acute steady-state relationships among PCO2, plasma bicarbonate concentration, and plasma hydrogen concentration during graded degrees of acute hypercapnia.11,12 These observations were obtained by sequentially exposing unanesthetized normal human volunteers to increasing concentrations of inspired carbon dioxide in a large environmental chamber. Increasing degrees of hypercapnia are associated with a curvilinear rise in plasma bicarbonate concentration, with higher levels of PCO2 resulting in lesser incremental changes in bicarbonate concentration. This acute rise in bicarbonate is largely due to the chloride shift as described earlier. As a result of the modest increment in bicarbonate, the average rise in plasma [H+] is limited to 0.75 nEq/L per mm Hg rise in PCO2 rather than the 1 nEq/mm Hg rise that would have occurred if the plasma bicarbonate concentration did not change.13
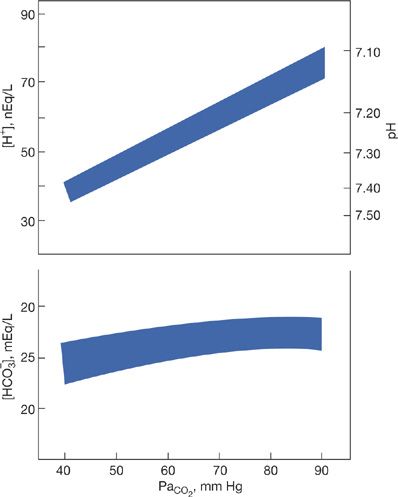
Figure 17-3 Ninety-five percent confidence bands for plasma hydrogen ion and bicarbonate concentrations during acute hypercapnia in normal humans. (Adapted with permission from Brackett NC Jr, Cohen JJ, Schwartz WB. Carbon dioxide titration curve of normal man: effect of increasing degrees of acute hypercapnia on acid-base equilibrium. New Engl J Med. 1965;272:6–12.)
The quantitative aspects of the adaptive response to acute hypercapnia are influenced markedly by the baseline acid–base status. Acute hypercapnia induces a larger increment in both plasma bicarbonate and H+ ion concentrations in animals with pre-existing hypobicarbonatemia (whether from metabolic acidosis or from chronic respiratory alkalosis) than in animals with pre-existing hyperbicarbonatemia (whether from metabolic alkalosis or from chronic respiratory acidosis).14 This points out that the factor controlling the amount of bicarbonate generated from an acute rise in PCO2 is not only the initial pH but also the initial bicarbonate concentration. Although the rise in bicarbonate in response to hypercapnia limits the fall in pH acutely, to excrete the gain of H+ produced from the rise in PCO2 requires renal compensatory mechanisms.
During the initial period of respiratory acidosis, renal compensation takes about 3 to 5 days, during which time there is enhanced reabsorption of proximal tubular bicarbonate, enhanced secretion of H+, and increased ammonia production.15
These processes will lead to an increase of the serum bicarbonate concentration and a rise in the systemic pH toward normal. However, when a steady state is achieved and a stable PCO2 is present, there is no longer an increase in ammonia production. As filtered bicarbonate is increased, there is enhanced proximal secretion of H+ and a normalization of intracellular pH removing the stimulus for ammonia synthesis.15
RENAL ADAPTATION TO RESPIRATORY ALKALOSIS
The adaptive responses to respiratory alkalosis occur in two distinct steps, in close analogy with respiratory acidosis. Hypocapnia reduces the carbonic acid concentration and causes a prompt fall in H+.16 Acutely, this alkalemia is ameliorated by a secondary, adaptive reduction in plasma bicarbonate concentration that stems principally from titration of nonbicarbonate body buffers.17 During protracted hypocapnia, renal adaptive mechanisms yield a further and larger secondary reduction in plasma bicarbonate that results in still greater amelioration of the alkalemia.17
In acute uncomplicated respiratory alkalosis the plasma bicarbonate concentration falls by approximately 0.2 mEq/L for each mm Hg reduction in PCO2. Thus, a reduction in plasma bicarbonate of 3 to 4 mEq/L occurs within minutes after PCO2 is lowered to 20 to 25 mm Hg. The resulting change in plasma H+ concentration is approximately 0.75 mEq/L for each mm Hg fall in PCO2, similar to the relationship between PCO2 and H+ in acute hypercapnia.
When hypocapnia persists beyond the acute phase, the additional decrement in plasma bicarbonate concentration is a consequence of renal adaptive responses and reflects a dampening of hydrogen ion secretion by the renal tubule.5 As a result, a transient suppression of net acid excretion occurs, largely manifested by a fall in ammonium excretion and by an increase in net bicarbonate excretion. These changes lead, in turn, to a positive hydrogen ion balance and a reduction in the body’s bicarbonate stores. Persistence of the resulting hypobicarbonatemia is explained by the continued inhibition of tubular hydrogen ion secretion and suppression of bicarbonate reabsorption.
The adaptive retention of acid during chronic hypocapnia is normally accompanied by a loss of sodium into the urine; the resultant decrease in the extracellular volume promotes chloride retention and the typical hyperchloremia of chronic respiratory alkalosis.18 Upon reaching a new steady state, the net excretion of acid returns to control levels, and the altered anionic concentration of the extracellular fluid (ECF), namely hypobicarbonatemia and hyperchloremia, is maintained by a reduced bicarbonate reabsorption and enhanced chloride reabsorption. On average, the combined effect of cell buffers and renal compensation results in a new steady state in which the plasma HCO3– concentration falls approximately 4 mEq/L for each 10 mm Hg reduction in the PCO2.19 The renal adaptation to persistent hypocapnia appears to be mediated by some direct effect of PCO2 itself, not the systemic pH. In animals in which plasma bicarbonate was reduced by HCl loading prior to adaptation to sustained hypocapnia, the renal response to a primary reduction in PCO2 was the same as in normal individuals, even though the net effect of this adaptation was an overt fall in pH.
RESPIRATORY ADJUSTMENT TO METABOLIC ACIDOSIS
Metabolic acidosis stimulates both central and peripheral chemoreceptors to increase alveolar ventilation and decrease PCO2 to limit the fall in pH. Although peripheral chemoreceptors appear to play a small role, in animal experiments the same degree of respiratory compensation occurs with intact and with ablated peripheral chemoreceptors. The increase in ventilation begins within 1 to 2 hours and reaches its maximal level at 12 to 24 hours. The stereotype is Kussmaul’s breathing in acute diabetic ketoacidosis, in which tidal volume is characteristically large with minute ventilation increasing by as much as 35 L. On average, studies in otherwise normal patients with metabolic acidosis reveal that the PCO2 will fall 1.2 mm Hg for every 1.0 mEq/L reduction in plasma HCO3– down to a minimum PCO2 of 10 to 15 mm Hg.20
On the other hand, failure to mount the expected ventilatory response to metabolic acidosis is an important indicator of respiratory decompensation. Daniel et al.21 in 140 critically ill trauma patients with metabolic acidosis applied the traditional formula derived from patients with chronic metabolic acidosis. Those whose PaCO2 exceeded the predicted PaCO2 by 2 mm Hg or more were 4.2 times more likely to be intubated and compensation status was an independent predictor of intubation as early as 60 minutes after episodes of significant hypotension.
RESPIRATORY ADJUSTMENT TO METABOLIC ALKALOSIS
The development of metabolic alkalosis is sensed by the respiratory chemoreceptors resulting in a decline in alveolar ventilation and an elevation of the PCO2. On average, the PCO2 rises 0.7 mm Hg for every 1.0 mEq/L increment in the plasma HCO3– concentration.18 Values significantly different from the predicted value represent superimposed respiratory acidosis or alkalosis. However, it is unclear whether this response significantly protects the pH from rising. In experimental animals, the rise in PCO2 in metabolic alkalosis increases net H+ excretion leading to an increase in the HCO3– concentration. The effect after several days is that the arterial pH is the same as it would have been if there had been no respiratory compensation.6,18
Ventilation may be strongly affected by influences other than acid–base balance. Among these influences are body temperature, increases in circulating catecholamines, changes in cerebral blood flow, changes in systemic blood pressure, and changes in metabolic activities of different organs (e.g., liver), as well as the physiologic state of the lung itself. Perhaps for teleologic reasons, the defense of chronic metabolic acid–base imbalances by ventilatory compensation is not of major importance.
ALTERNATIVE CONCEPTS OF ACID–BASE BALANCE
The preceding discussion has tacitly assumed that the systemic pH is the final control that affects the renal and respiratory response to an acid–base disorder; however, this issue is certainly not settled. The proximal tubular cell of the kidney can often have effects that are more predictably based on the PCO2 rather than the arterial pH. If PCO2 is elevated, the proximal tubular cells act to secrete protons and reabsorb bicarbonate whether or not there is systemic alkalosis or acidosis. This may be explained if an elevation in PCO2 results in intracellular acidosis and the cell is responding appropriately to its internal milieu.6,18 Similarly, in the central control of respiration, it is controversial as to whether it is CSF pH, interstitial pH, PCO2, or the bicarbonate concentration that stimulates compensatory changes in ventilation.7
In addition to the previously mentioned observations, it is also known that changes in salt and water balance may affect acid–base status. For example, Schwartz’s group18 found that a low dietary sodium chloride intake in dogs with a stable amount of water intake results in hypoventilation, increased PCO2, and increased HCO3– concentration. Studies in dogs have demonstrated that increasing dietary NaCl with a fixed water intake increases the acidity of body fluids, whereas decreasing the NaCl in diet with a fixed water intake decreases the acidity of body fluids.22
An alternative view to understanding acid–base disorders and the regulatory response of the lungs and kidneys is offered by the theories initially proposed by Stewart.23,24 Based on physicochemistry, Stewart emphasized the important principle that H+ and HCO3– as well as the acidic and anionic forms of weak acids are actually dependent variables in a solution. The three independent variables, PCO2, the strong ion difference (SID), and the total weak anion concentration, can be manipulated externally and serve to determine the concentration of the dependent variables, H+ and HCO3–. The major components of the weak anions in plasma are the albumin and inorganic phosphate concentrations. The SID is the difference between the sums of all strong cations and all strong anions:
![]()
This equation is based on the principles of: (1) electroneutrality, (2) dissociation equilibria of all incompletely dissociated substances, and (3) conservation of mass. This concept appears to better explain the basis for renal and ventilatory response in a variety of states which also affects acid–base balance. Practically, it is observed that the plasma SID is primarily regulated by the kidneys, whereas the PCO2 is regulated by alveolar ventilation. The weak anion concentration is generally not regulated and may often be assumed to be stable.
This concept has primarily been used by investigators in relation to the study of central regulation of ventilation.7 As albumin and other proteins are not present in the CSF, it is the SID and PCO2 that determine the concentration of weakly dissociating electrolytes, H+, OH–, and HCO3–. In analyzing various acid–base disturbances, it appears that the change in CSF SID can predict the concentration of CSF bicarbonate.7
In evaluating acid–base balance in many species, there is a consistent inverse relationship between the pH and body temperature, whereas the CO2 content remains stable.4,25 To explain this relationship Reeves and his coworkers provided evidence that the imidazole ring structure of histidine is responsible for the pH–temperature relationship.26 This is because imidazole has a pKa in the physiologic range (7.00), is relatively ubiquitous, and has total energy of ionization (7 kcal/mol). To integrate acid–base regulation with receptor function and control of respiration, Reeves and Rahn4 have proposed the hypothesis that it is not the arterial or intracellular pH that is being regulated per se but rather the constancy of the fractional dissociation of the imidazole moiety of histidine contained in proteins throughout the body.
α-Imidazole is defined as the ratio of the absolute amount of unprotonated imidazole (Im) to total imidazole (HIm + Im):
![]()
α-Imidazole regulation (alphastat regulation) would have the effect of maintaining cellular protein charge states and enzymatic functions constant. It would also maintain the OH–/H+ ratio constant in all compartments. There is also evidence that alphastat regulation directly influences ventilatory status. For example, application of an imidazole blocker to the chemosensitive area of the medulla in cats blocked increases in ventilation caused by local application of acid.27 Thus, changes in PCO2, reflecting alveolar ventilation, may be determined by alphastat regulation, which maintains the OH–/H+ ratio constant in membranes of the cells in the chemosensitive areas of the medulla.
The difficulty with using these concepts lies in the practical measurement of the relevant molecules. For example, although the imidazole moiety of histidine is considered the most important of the intracellular buffers,26 its pKa and total energy of ionization may vary widely due to the influence of the local configuration of molecules into which they are incorporated. Thus, even in lower animals such as fish under different temperatures, calculations based on the alphastat model do not accurately predict the acid–base disturbance, since the pKa and enthalpy of ionization vary with temperature and are difficult to measure.
Similarly, measurement of the plasma SID is problematic and is often replaced by the “SID effective,” which is roughly equal to the bicarbonate concentration plus albumin and inorganic phosphate.23 Calculation of the anion gap ![]() – accounts for the roles of the strong ions Na+ and Cl– as well as bicarbonate but does not account for the role of inorganic phosphate or plasma proteins. Although the bicarbonate concentration may not be, strictly speaking, an independent variable, the AG calculation does indicate the quantity of unmeasured anions and hence is an indirect measure of the SID. If one considers the impact of serum proteins and inorganic phosphate in the unmeasured anion pool, the AG gives a very useful parameter in evaluating acid–base disturbances. Many studies have compared the utility of SID versus corrected AG (corrected for the level of serum albumin) and found that it was no more accurate in making a diagnosis. One study of patients in an intensive care unit, reported that Stewart’s method diagnosed underlying metabolic acidosis in 22 patients (of the total of 152 patients in the study) with normal plasma bicarbonate level. However, when the AG was corrected for hypoalbuminemia, it was elevated in all of the samples with normal bicarbonate showing the effectiveness of the traditional approach.28 In another study of 935 ICU patients, the Stewart method detected metabolic acidosis in 14% patients with normal bicarbonate levels, whereas the traditional method made a similar diagnosis in 13% of patients.29 A recent study in patients with septic shock and liver transplantation found an excellent correlation between SID and corrected AG.30
– accounts for the roles of the strong ions Na+ and Cl– as well as bicarbonate but does not account for the role of inorganic phosphate or plasma proteins. Although the bicarbonate concentration may not be, strictly speaking, an independent variable, the AG calculation does indicate the quantity of unmeasured anions and hence is an indirect measure of the SID. If one considers the impact of serum proteins and inorganic phosphate in the unmeasured anion pool, the AG gives a very useful parameter in evaluating acid–base disturbances. Many studies have compared the utility of SID versus corrected AG (corrected for the level of serum albumin) and found that it was no more accurate in making a diagnosis. One study of patients in an intensive care unit, reported that Stewart’s method diagnosed underlying metabolic acidosis in 22 patients (of the total of 152 patients in the study) with normal plasma bicarbonate level. However, when the AG was corrected for hypoalbuminemia, it was elevated in all of the samples with normal bicarbonate showing the effectiveness of the traditional approach.28 In another study of 935 ICU patients, the Stewart method detected metabolic acidosis in 14% patients with normal bicarbonate levels, whereas the traditional method made a similar diagnosis in 13% of patients.29 A recent study in patients with septic shock and liver transplantation found an excellent correlation between SID and corrected AG.30
Thus, as will be described in more detail in the following section, the use of the AG is still the most clinically useful tool to determine the contribution of different metabolic etiologies of metabolic acidosis.
APPROACH TO PATIENTS WITH AN ACID–BASE DISTURBANCE
In this section, we examine the diagnostic approach to disorders of acid–base balance with a particular emphasis on the ventilatory response and its role in mitigating or exacerbating acid–base disorders. We will also review the approach to the patient with complex acid–base disorders.
 ANALYSIS OF CLINICAL INFORMATION
ANALYSIS OF CLINICAL INFORMATION
Table 17-1 summarizes the pattern of abnormality of arterial blood acid–base parameters in the four classic acid–base disorders. It also indicates the physiologic or compensatory response induced in pulmonary or renal function in response to the initial disturbance.
Base Excess and Base Deficit Notations
Base excess and base deficit are terms applied to an analytic method for determination of the appropriateness of responses to disorders of acid–base metabolism.31 The base excess or deficit is determined by measuring blood pH against ambient PCO2 and against a PCO2 of 40 mm Hg. If the calculated HCO3– is below 25 when the PCO2 is 40 mm Hg and the original pH is low, a base deficit is indicated. The magnitude of the deficit is expressed as the number of mEq of bicarbonate needed to restore the serum bicarbonate to 25 mEq/L at a PCO2 of 40 mm Hg compared to that at the ambient PCO2. The use of notations for base excess and deficit has been debated in the medical literature. This notation is favored in the evaluation of acid–base status in the operating room because acute changes in PCO2 and in HCO3– can be simply evaluated by this approach. However, this notation can be misleading in chronic respiratory alkalosis or acidosis, since the patient with chronic respiratory alkalosis will be categorized as suffering from a base deficit because of the low serum bicarbonate induced as compensation for the reduced PCO2. In fact, a “base deficit” is a normal physiologic response to the chronic reduction in PCO2. Unfortunately, lack of familiarity with the complete analytical paradigm used for this analysis of acid–base disorders has led some to focus on the designations “base deficit” and “base excess” as guides to bicarbonate or acid therapy in chronic respiratory disorders. In addition, discrepancies between the buffering characteristics of plasma, blood, and whole body have also been cited as potential weaknesses in a system for assessing acid–base disorders which relies on in vitro CO2 titration methods. We, therefore, recommend that the physiologic evaluation of the patient be the mode of analysis of acid–base disorders rather than an emphasis on derived formulae.
Use of Nomograms
As indicated earlier, the body buffers and the kidneys respond in a predictable fashion to a change in PCO2 whereas ventilatory response to changes in [HCO3–] is also predictable. Also, the resulting changes in bicarbonate and pH are time dependent so that a larger change occurs in several days than in the first hours. The confidence bands for changes in PCO2 or HCO3– in response to primary disturbances are shown in Figure 17-4.7 Any deviation can be interpreted as a reflection of processes other than a compensatory response. For example, in a patient with chronic obstructive airways disease, other factors affecting the acid–base status are the concentration of potassium in the plasma, the size of ECF volume, chloride depletion, diuretics, renal hypoperfusion, and coexisting renal disease. The special case of posthypercapnic alkalosis is discussed in the next section.
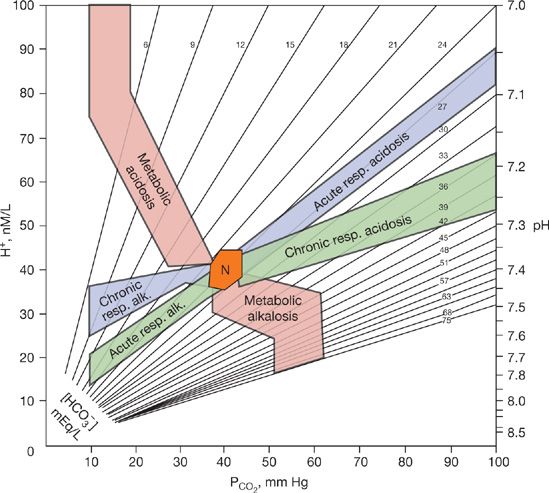
Figure 17-4 Acid–base map showing the normal range (N) and the confidence bands for acute or chronic respiratory and metabolic acid–base disturbances. The ordinates are the partial pressure of CO2 and the hydrogen-ion activity given in nmol/L and pH units. Isopleths for bicarbonate concentration, in mEq/L, are also shown. (Reproduced with permission from Goldberg M, Green SB, Moss ML, et al. Computer-based instruction and diagnosis of acid-base disorders. A systematic approach. JAMA. 1973;223(3):269–278.)
In evaluating an acid–base disorder, the history and physical examination are invaluable in focusing attention on potential pathologic processes.32 The composition of blood, with respect to serum electrolytes and blood gases, is then examined for consistency with clinical impressions. However, in using the acid–base map (Fig. 17-4), remember that the map is based on data from individuals who had a single disorder. Therefore, the map does not take into account the possibility of multiple disorders. For example, in a patient with chronic obstructive airways disease whose sputum has turned purulent and who develops nausea and vomiting, the possibility arises of coexistent metabolic alkalosis and acute respiratory acidosis. However, ill-advised application of the arterial blood-gas values from this patient (e.g., pH = 7.25 and PCO2 = 75 mm Hg) to the acid–base map would lead to the erroneous conclusion that a chronic respiratory acidosis is present. Thus, the clinician needs to integrate laboratory data with clinical assessments to properly analyze clinical disorders of acid–base balance.
 APPROACH TO THE PATIENT WITH METABOLIC ACIDOSIS
APPROACH TO THE PATIENT WITH METABOLIC ACIDOSIS
An increase in the H+ concentration of the ECF will result in a series of predictable responses which allow the clinician to ascertain the appropriateness of organized homeostatic responses to the perturbation.20 The pathophysiologic basis for the initiation of metabolic acidosis and homeostatic responses in the defense of systemic pH have been defined earlier in descriptions of the buffering of newly introduced acid [see Eq. (1)] and in the demonstration of the normal confidence band for the ventilatory response to metabolic acidosis as detailed in the acid–base nomogram (Fig. 17-4).
A key clinical distinction in the pathogenesis of metabolic acidosis is whether the production of the acidosis is rapid or slow. If the etiology of the metabolic acidosis is merely the continued ingestion of a diet which generates a variety of fixed acids such as H2SO4 [see Eq. (2)] from the metabolism of methionine residues, then the serum HCO3– will fall slowly as only that fraction of the 50 to 100 mEq of H+ generated from diet that is not excreted would be added to the body fluids each day. However, if the addition occurs because of an acute increase in the acid load such as may occur with lactic acidosis, the kidney capacity can be rapidly overwhelmed, and serum bicarbonate may fall precipitously. See Table 17-2 for the common causes of metabolic acidosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


