CARDIOMYOPATHY AND MYOCARDITIS
Lynne Warner Stevenson ![]() Joseph Loscalzo
Joseph Loscalzo
DEFINITION AND CLASSIFICATION
Cardiomyopathy is a disease of the heart muscle. It is estimated that cardiomyopathy accounts for 5–10% of the 5–6 million patients already diagnosed with heart failure in the United States. This term is intended to exclude cardiac dysfunction that results from other structural heart disease, such as coronary artery disease, primary valve disease, or severe hypertension; however, in general usage the phrase ischemic cardiomyopathy is sometimes applied to describe diffuse dysfunction occurring in the presence of multivessel coronary artery disease, and nonischemic cardiomyopathy to describe cardiomyopathy from other causes. As of 2006, cardiomyopathies are defined as “a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic.”1
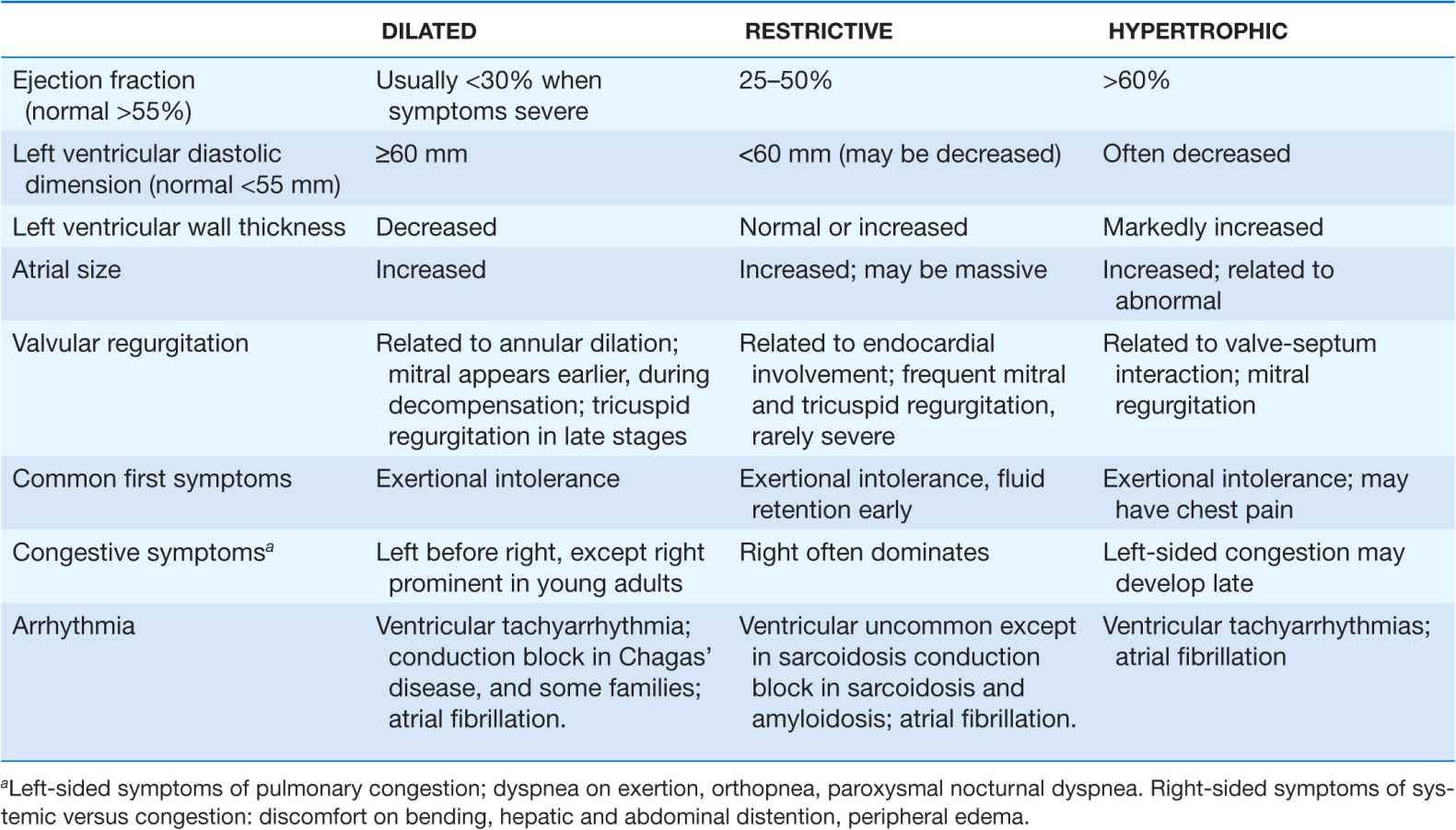
The traditional classification of cardiomyopathies into a triad of dilated, restrictive, and hypertrophic was based initially on autopsy specimens and later on echocardio-graphic findings. Dilated and hypertrophic cardiomyopathies can be distinguished on the basis of left ventricular wall thickness and cavity dimension; however, restrictive cardiomyopathy can have variably increased wall thickness and chamber dimensions that range from reduced to slightly increased, with prominent atrial enlargement. Restrictive cardiomyopathy is now defined more on the basis of abnormal diastolic function, which is also present but initially less prominent in dilated and hypertrophic cardiomyopathy. Restrictive cardiomyopathy can overlap in presentation, gross morphology, and etiology with both hypertrophic and dilated cardiomyopathies (Table 21-1).
TABLE 21-1
PRESENTATION WITH SYMPTOMATIC CARDIOMYOPATHY
Expanding information renders this classification triad based on phenotype increasingly inadequate to define disease or therapy. Identification of more genetic determinants of cardiomyopathy has suggested a four-way classification scheme of etiology as primary (affecting primarily the heart) and secondary to other systemic disease. The primary causes are then divided into genetic, mixed genetic and acquired, and acquired; however, in current practice the genetic information is often unavailable at the time of initial presentation, particularly in the absence of extracardiac manifestations. Many mutated genes can be associated with the same general phenotype, and one defective gene may manifest as multiple phenotypes. In addition, the bases of evidence for most therapies are still driven by clinical phenotypes. Although the proposed genetic classification does not yet guide many current clinical strategies, it will become increasingly relevant as classification of disease moves beyond individual organ pathology to more integrated systems approaches.
GENERAL PRESENTATION
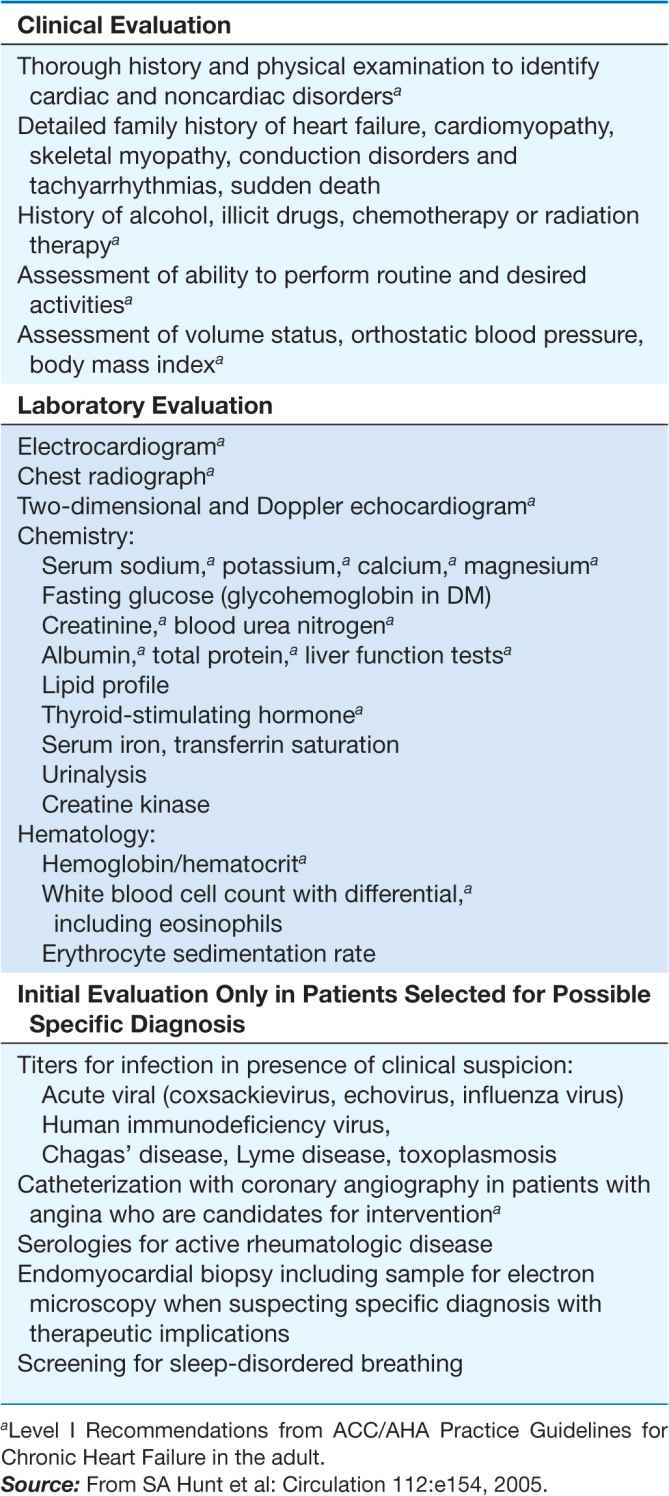
For all cardiomyopathies, the early symptoms often relate to exertional intolerance with breathlessness or fatigue, usually from inadequate cardiac reserve during exercise. These symptoms may initially go unnoticed or be attributed to other causes, commonly pulmonary. As fluid retention leads to elevation of resting filling pressures, shortness of breath may occur during routine daily activity, such as dressing, and may manifest as dyspnea or cough in the supine position. Although often considered the hallmark of congestion, peripheral edema may not appear despite severe fluid retention, particularly in younger patients. The nonspecific term congestive heart failure describes only the resulting syndrome of fluid retention, which is common to the three types of cardiomyopathy and also to other cardiac diseases associated with elevated filling pressures. Despite the different structural basis, all three types of cardiomyopathy can be associated with atrioventricular valve regurgitation, typical and atypical chest pain, atrial and ventricular tachyarrhythmias, and embolic events (Table 21-1). Initial evaluation begins with a detailed clinical history and examination, looking for clues to cardiac, extracardiac, and familial disease (Table 21-2). The initial evaluation, prognosis, and therapy are generally defined by the severity of cardiac and clinical dysfunction, with some distinctive features according to etiology.
TABLE 21-2
INITIAL EVALUATION OF CARDIOMYOPATHY
GENETIC ETIOLOGIES OF CARDIOMYOPATHY
The estimated prevalence of a genetic etiology for cardiomyopathy continues to increase with increasing awareness of the importance of the family history and the availability of genetic testing. Well recognized in hypertrophic cardiomyopathy, heritability is present in at least 30% of dilated cardiomyopathies without other clear etiology. Careful family history should elicit not only known cardiomyopathy and heart failure, but also family members who have had sudden death, often incorrectly attributed to “a massive heart attack,” who have had atrial fibrillation or pacemaker implantation by middle age, or who have muscular dystrophy. The family history should be reviewed at subsequent intervals particularly regarding siblings and cousins, who may tend to manifest disease at similar ages.
Most familial cardiomyopathies are inherited in an autosomal dominant pattern, with occasional autosomal recessive and X-linked inheritance (Table 21-3). The penetrance and phenotype of a given mutation varies with other genetic, epigenetic, and environmental determinants. Some mutations are associated with primary conduction system disease as well as dilated cardiomyopathy (CDDC). With rare exceptions, such as the replacements for defective metabolic enzymes, current therapy is based on the phenotype rather than the genetic defect. However, knowledge of the genetic defect may influence prognosis and in some cases provide indication for implantable defibrillators.
Defects in sarcomeric proteins of myosin, actin, and troponin are the best characterized. While the majority of these are associated with hypertrophic cardiomyopathy, an increasing number of sarcomeric mutations have now been implicated in dilated cardiomyopathy, and some have also been associated with left ventricular noncompaction. Thus far, few mutations have been identified in excitation-contraction coupling proteins, perhaps because they are too crucial for survival to allow variation.
Many of the proteins encoded by abnormal structural genes span more than one functional area of the myocyte (Fig. 21-1). Proteins contributing to the Z-disk organize and stabilize the sarcomeres. Multiple other proteins are involved in connecting and maintaining the cytoskeleton of the myocyte. For example, desmin forms intermediate filaments that connect the nuclear and plasma membranes, Z-lines, and the intercalated disks between muscle cells. Desmin mutations impair the transmission of force and signaling for both cardiac and skeletal muscle, and are, thus, associated with a peripheral myopathy as well as a dilated cardiomyopathy. Most of the identified genetic defects in the Z-disk and cytoskeleton are associated with dilated cardiomyopathy.
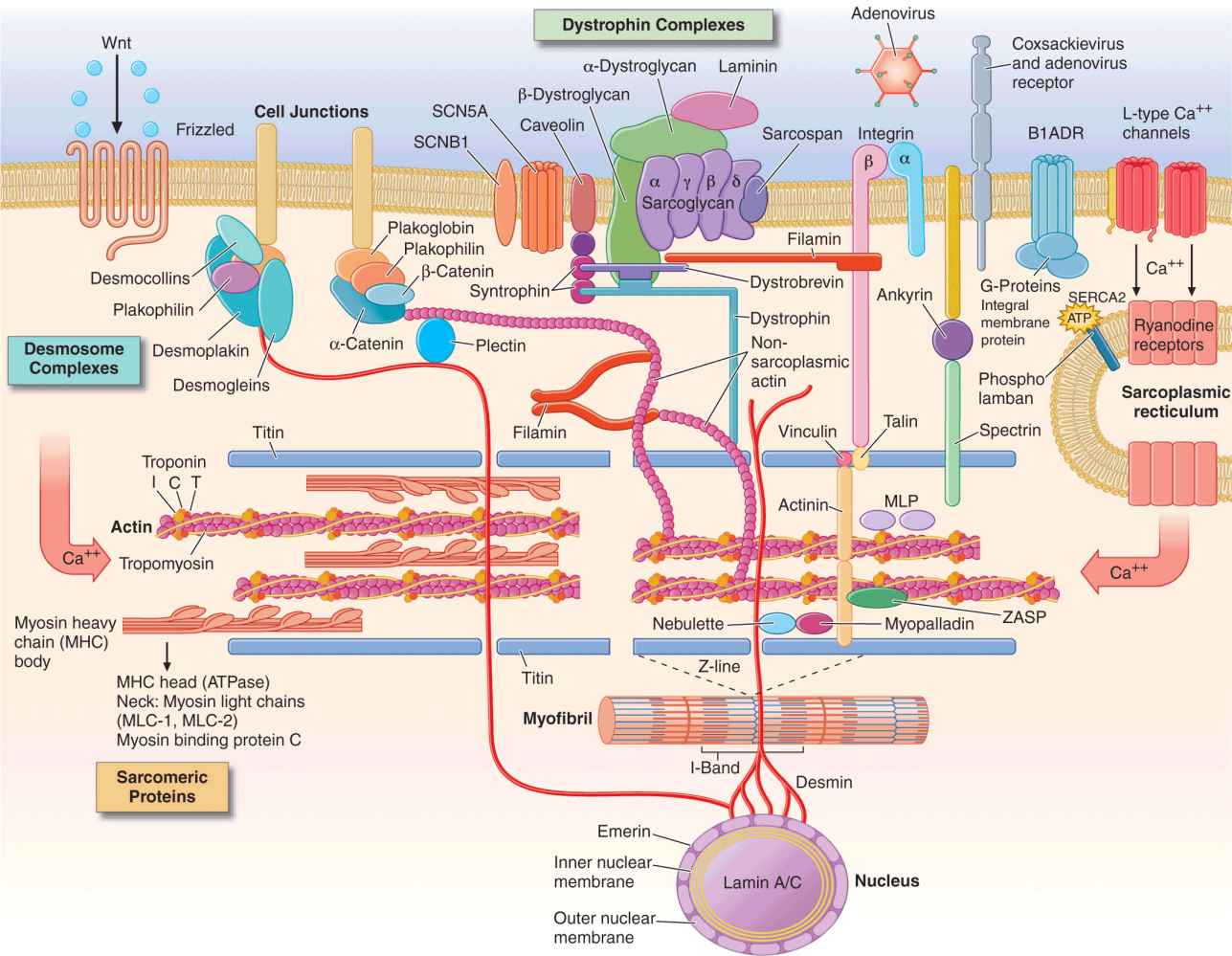
FIGURE 21-1
Drawing of myocyte indicating multiple sites of abnormal gene products associated with cardiomyopathy. Major functional groups include the sarcomeric proteins (actin, myosin, tropomyosin, and the associated regulatory proteins), the dystrophin complex stabilizing and connecting the cell membrane to intracellular structures, the desmosome complexes associated with cell-cell connections and stability, and multiple cytoskeletal proteins that integrate and stabilize the myocyte. ATP, adenosine triphosphate. (Figure adapted from Jeffrey A. Towbin, MD, University of Cincinnati, with permission.)
Proteins in the sarcolemmal membrane are associated with dilated cardiomyopathy. The best known is the X-linked dystrophin, abnormalities of which cause Duchenne’s and Becker’s muscle dystrophy. (Interestingly, abnormal dystrophin can be acquired when the coxsackievirus cleaves dystrophin during viral myocarditis.) This protein provides a network that supports the sarcolemma and also connects to the sarcomere. The progressive functional defect in both cardiac and skeletal muscle reflects vulnerability to mechanical stress. Dystrophin is associated at the membrane with a complex of other proteins, such as metavinculin, abnormalities of which cause dilated cardiomyopathy with autosomal dominant inheritance. Defects in the sarcolemmal channel proteins (channelopathies) are generally associated with primary arrhythmias, but mutations in SCN5A, distinct from those which cause the Brugada or long-QT syndromes, have been implicated in dilated cardiomyopathy.
Nuclear membrane protein defects in the myocyte can also cause skeletal myopathy in either autosomal dominant (lamin proteins) or X-linked (emerin) patterns. These are associated with a high prevalence of atrial arrhythmias and conduction system disease which, in some family members, occur without detectable cardiomyopathy.
Intercalated disks between cardiac myocytes allow mechanical and electrical coupling between cells and also connect to desmin filaments within the cell. Mutations in proteins of the desmosomal complex compromise attachment of the myocytes, which can become disconnected and die, to be replaced by fat and fibrous tissue. These areas are highly arrhythmogenic and may go on to aneurysm formation. Although it is more noticeable in the thinner right ventricle, this condition often affects both ventricles. As desmosomes are also important for elasticity of hair and skin, some defective desmosomal proteins are associated with striking “woolly hair” and thickened skin on the palms and soles.
Owing to the conservation of signaling pathways in multiple systems, we may expect to discover more extracardiac manifestations of genetic abnormalities initially considered to manifest exclusively in the heart. In contrast, the monogenic disorders of metabolism that affect the heart are already clearly recognized to affect multiple organ systems (Table 21-4). The most important currently are those defective enzymes for which specific enzyme replacement therapy can now ameliorate the course of disease, as with alpha-galactosidase-A (Fabry’s disease). Abnormalities of mitochondrial DNA (maternally transmitted) impair energy production with multiple clinical manifestations, including impaired cognitive function and skeletal myopathy. The phenotypic expression is highly variable depending on the distribution of the maternal mitochondria during embryonic development. Heritable systemic diseases, such as familial amyloidosis and hemochromatosis, can affect the heart without abnormal expression of specific cardiac genes.
For any patient with suspected or proven genetic disease, family members should be considered and evaluated in a longitudinal fashion. Screening includes an echocardiogram and electrocardiogram (ECG). The indications and implications for confirmatory specific genetic testing vary depending upon the specific mutation. The profound questions raised by families about diseases shared and passed down merit serious and sensitive discussion, ideally provided by a trained genetic counselor.
DILATED CARDIOMYOPATHY
An enlarged left ventricle with decreased systolic function as measured by left ventricular ejection fraction characterizes dilated cardiomyopathy (Figs. 21-2, 21-3, and 21-4). Systolic failure is more marked than the frequently accompanying diastolic dysfunction, although the latter may be functionally severe in the setting of marked volume overload. The syndrome of dilated cardiomyopathy has multiple etiologies (Table 21-5). Up to one-third of cases may be familial, as discussed later. Acquired cardiomyopathy is often attributed to a brief primary injury such as infection or toxin exposure. Some myocytes may die during the initial injury, while others survive only to have later programmed cell death, (apoptosis). As the surviving myocytes hypertrophy to accommodate the increased burden of wall stress, local and circulating factors stimulate deleterious responses that contribute to progression of disease, even in the absence of further primary injury. Dynamic remodeling of the interstitial scaffolding affects diastolic function and the amount of ventricular dilation. Mitral regurgitation commonly develops as the valvular apparatus is distorted by ventricular dilation and sometimes by focal injury to underlying myocardium and is usually substantial by the time heart failure is severe. Many cases that present “acutely” have progressed silently through these stages over months to years.
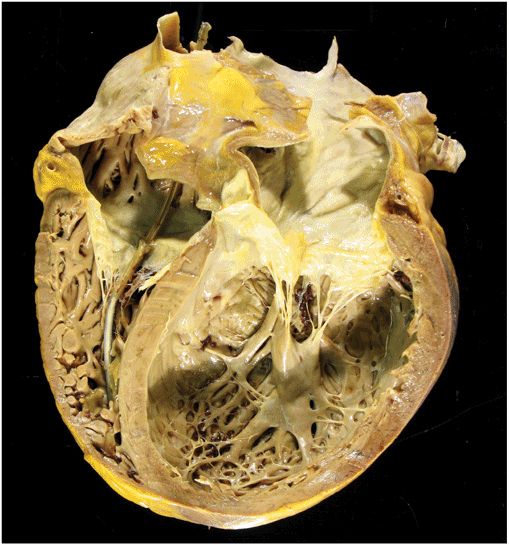
FIGURE 21-2
Dilated cardiomyopathy. This gross specimen of a heart removed at the time of transplantation shows massive left ventricular dilation and moderate right ventricular dilation. Although the left ventricular wall in particular appears thinned, there is significant hypertrophy of this heart, which weighs more than 800 gm (upper limit of normal = 360 g). A defibrillator lead is seen traversing the tricuspid valve into the right ventricular apex. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
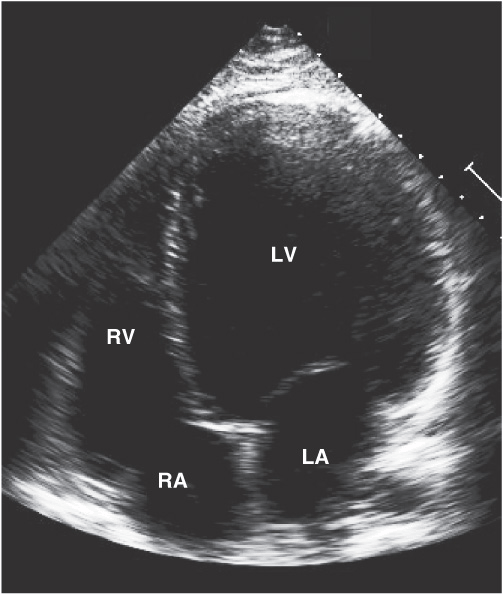
FIGURE 21-3
Dilated cardiomyopathy. This echocardiogram of a young man with dilated cardiomyopathy shows massive global dilation and thinning of the walls of the left ventricle (LV). The left atrium (LA) is also enlarged compared to normal. Note that the echocardiographic and pathologic images are vertically opposite, such that the LV is by convention on the top right in the echocardiographic image and bottom right in the pathologic images. (Image courtesy of Justina Wu, MD, Brigham and Women’s Hospital, Boston.)
Regardless of the nature and degree of direct cell injury, the resulting functional impairment often includes some contribution from secondary responses that may be reversible. The potential reversibility of cardiomyopathy in the absence of ongoing injury remains a subject of active controversy. Almost half of all patients with truly recent onset cardiomyopathy demonstrate substantial spontaneous recovery. Some patients have dramatic improvement to near-normal ejection fractions during pharmacologic therapy, particularly notable with the β-adrenergic antagonists coupled with renin-angiotensin system inhibition. Interest in the potential for recovery of cardiomyopathy in the absence of coronary artery disease has been further stimulated by occasional “recovery” of left ventricular function in young patients after a year or more of mechanical circulatory support. The diagnosis and therapy for dilated cardiomyopathy is generally dictated by the stage of heart failure (Chap. 17), with specific aspects discussed with the relevant etiology later in this chapter.
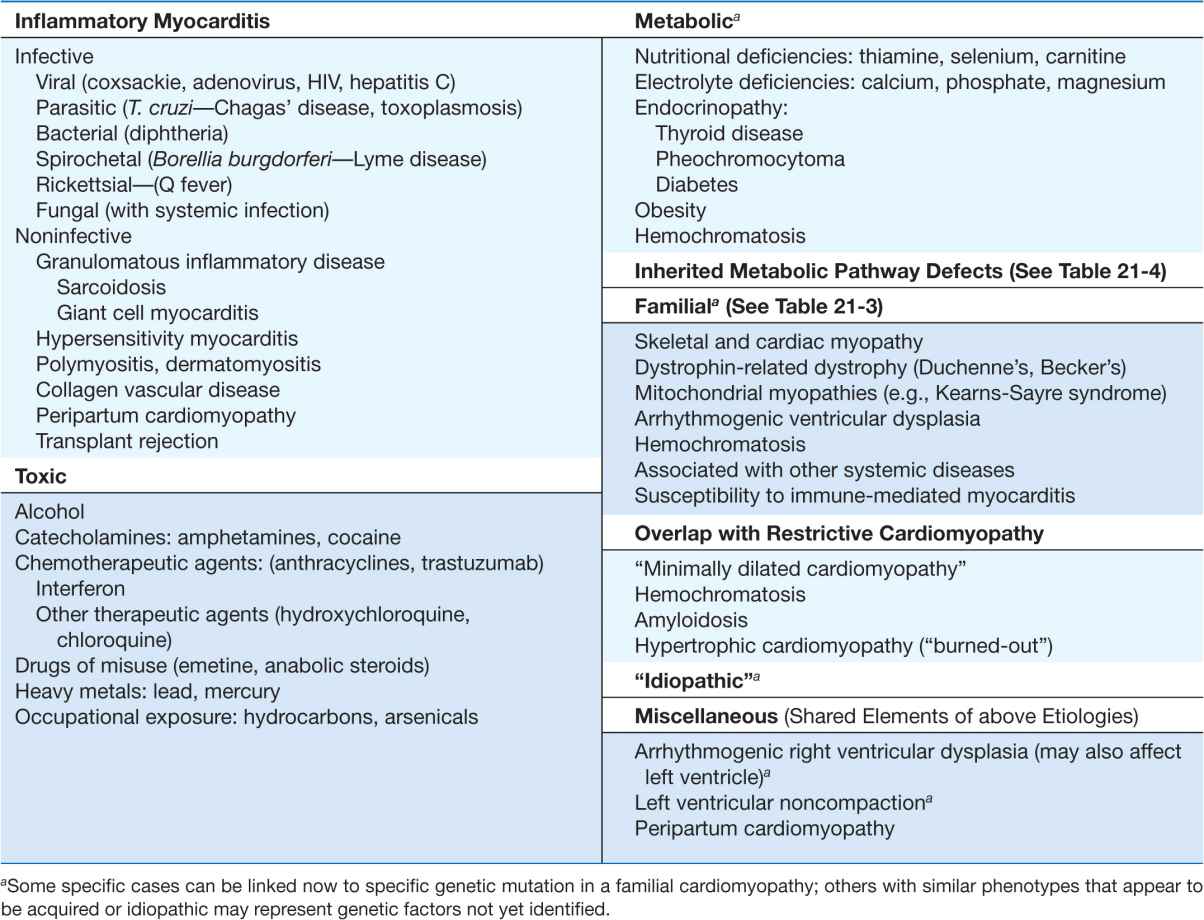
TABLE 21-5
MAJOR CAUSES OF DILATED CARDIOMYOPATHY (WITH COMMON EXAMPLES)
INFECTIVE MYOCARDITIS
Myocarditis is an inflammatory process, most commonly attributed to infectious organisms that can invade the myocardium directly, produce cardiotoxins, and trigger chronic inflammatory responses. Infective myocarditis has been reported with almost all types of infectious agents, but is most commonly associated with viral infections, the protozoan Trypanosoma cruzi in South America, and endomyocardial fibrosis in equatorial Africa.
Viral myocarditis in murine models begins with acute infection. After viruses enter the circulation through the respiratory or gastrointestinal tract, they can infect other organs possessing specific receptors, such as the coxsackie-adenovirus receptor on the heart. Viral invasion and replication can lead directly to myocardial injury and lysis. Viral proteases have multiple actions, of which one is to degrade the protein, dystrophin, in the myocyte membrane complex that is genetically abnormal in some muscular dystrophies. Viral antigens activate immune responses that help to contain the initial infection but may persist into later phases. Components include nonspecific cytokines, specific antibodies, and cytotoxic T-lymphocytes, which in some cases recognize myocyte proteins. There is varying evidence for a latent phase of ongoing infection with persistence of the viral genome and some viral proteins. The relative contributions of viral persistence and deleterious host immune responses to progressive dysfunction have not been clearly delineated in human disease (Fig. 21-5). The late stages are dominated by nonspecific secondary changes in gene expression, and by local and systemic neurohormonal responses, as seen for other etiologies of heart failure.
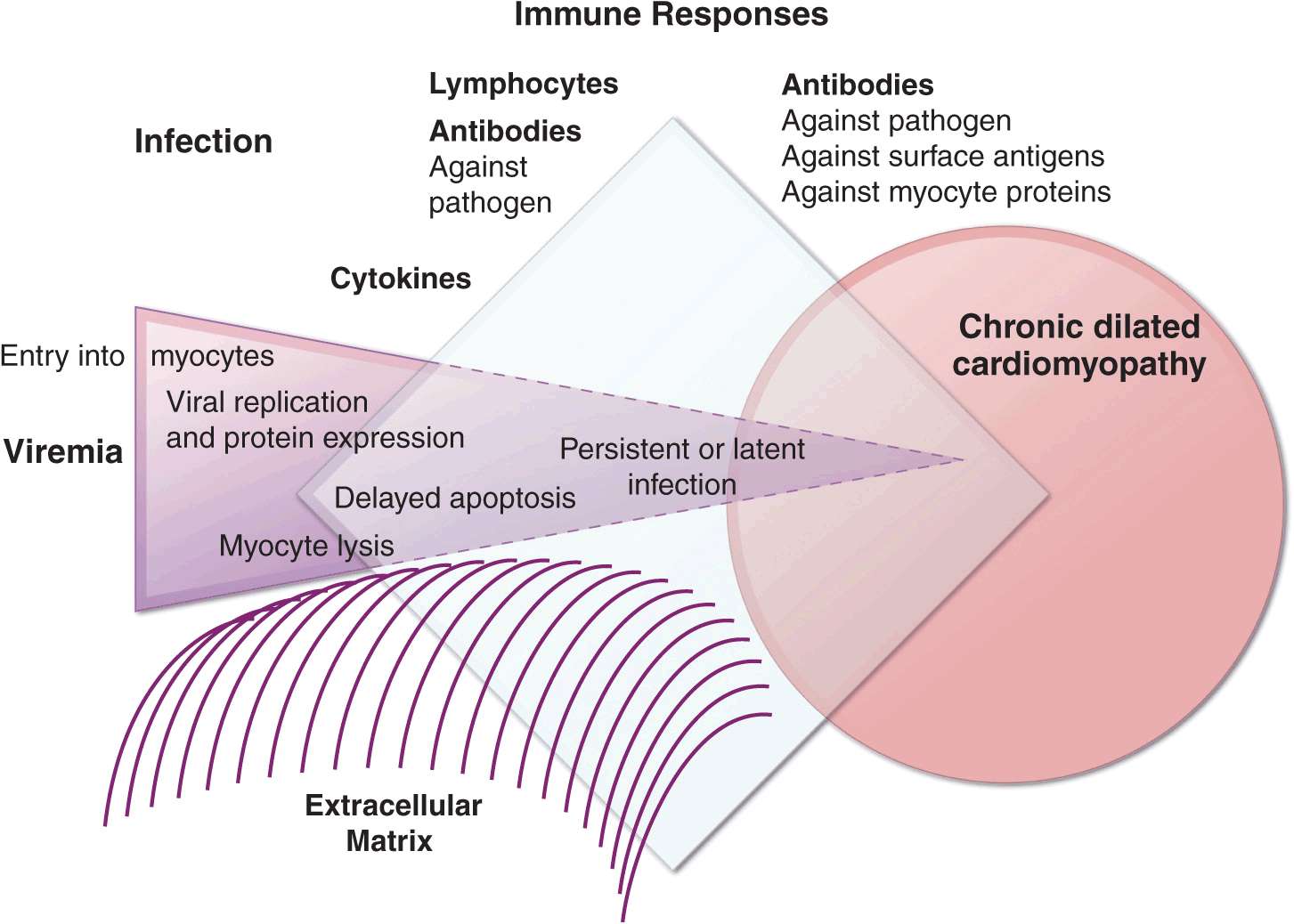
FIGURE 21-5
Schematic diagram demonstrating the possible progression from infection through direct, secondary, and autoimmune responses to dilated cardiomyopathy. Most of the supporting evidence for this sequence is derived from animal models. It is not known to what degree persistent infection and/or ongoing immune responses contribute to ongoing myocardial injury in the chronic phase.
Although viral myocarditis is generally considered to be an acquired cardiomyopathy, families have been reported whose clinical disease appeared after a syndrome consistent with viral myocarditis. One possible explanation for this apparent mixed etiology is that some genetic variants of myocardial cell surface receptors bind more avidly to certain viruses, particularly coxsackievirus and adenovirus.
The typical clinical picture of myocarditis is a young adult with progressive dyspnea and weakness over a few days to weeks after a recent viral syndrome with fevers and often myalgias indicative of skeletal muscle inflammation. Some patients present with atypical or anginal-type chest pain, or with pleuritic, positional chest pain due to pericarditis with some degree of underlying myocarditis. Patients in whom ventricular tachyarrhythmias dominate the presentation may have viral myocarditis but should be evaluated for sarcoidosis or giant cell myocarditis. Patients presenting with pulmonary or systemic embolic events from intracardiac thrombi generally already have chronic, severe cardiac dysfunction.
A small number of patients present with acute fulminant myocarditis, with rapid progression from a severe febrile respiratory syndrome to cardiogenic shock from which multiple organ system failure, including coagulopathy, may develop. Such patients have often been discharged from the emergency department with antibiotic therapy only to return in extremis. Prompt triage is vital to provide aggressive support with high-level intravenous inotropic therapy and on occasion, mechanical circulatory support; importantly, more than half of patients with this acute presentation can survive with marked improvement within the first few weeks, often returning to near-normal systolic function.
Many patients presenting with heart failure after a viral illness actually have a long-standing cardiomyopathy that was acutely exacerbated but not caused by the new viral illness. Heart failure from any cause often worsens transiently during infection, presumably due to the myocardial depressant effects of circulating cytokines. Marked left ventricular dilation and the presence of severely elevated left-ventricular filling pressures without frank pulmonary edema suggest chronic, slowly progressive disease, which is often further supported by a history of gradual changes in exercise tolerance before the viral syndrome.
For the usual subacute presentation, the diagnosis of cardiomyopathy is confirmed by echocardiography, and further evaluation is directed to ascertain whether myocarditis is present. Troponin is often mildly elevated, and creatine kinase may be released from the cardiac injury or skeletal muscle involvement. In some cases, cardiac catheterization is performed to rule out acute ischemia. Magnetic resonance imaging is increasingly used for the diagnosis of myocarditis, which is supported by evidence of increased tissue edema and gadolinium enhancement, particularly in the mid-wall distinct from the usual coronary artery territories. Endomyocardial biopsy criteria for myocarditis require lymphocytic infiltration with evidence of myocyte necrosis (Fig. 21-6), but are met in only about 10–20% of classic presentations. Most biopsies in fulminant myocarditis show only marked tissue edema without a cellular infiltrate, and it is likely that many less acute cases may similarly be characterized by tissue edema and cytokine depression of myocardial function, possibly including some antibody-mediated endothelial injury, without marked cellular infiltrates. Acute and convalescent viral titers are usually sent but are more likely to be important from the public health standpoint than for the individual.
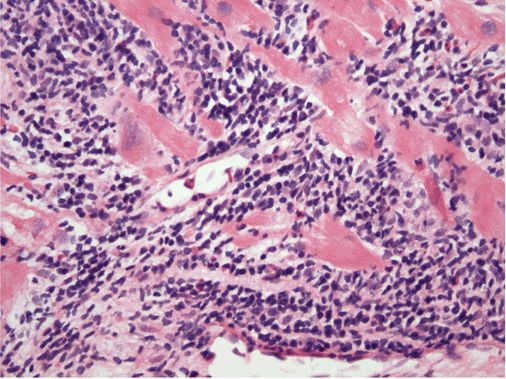
FIGURE 21-6
Acute myocarditis. Microscopic image of an endomyocardial biopsy showing massive infiltration with mononuclear cells and occasional eosinophils associated with clear myocyte damage. The myocyte nuclei are enlarged and reactive. Such extensive involvement of the myocardium would lead to extensive replacement fibrosis even if the inflammatory response could be suppressed. Hematoxylin and eosin stained section, 200× original magnification. (Image courtesy of Robert Padera, MD, PhD, Department of Pathology, Brigham and Women’s Hospital, Boston.)
Viral myocarditis treatment is initially directed toward stabilizing the hemodynamic status and then toward adjusting neurohormonal antagonists for the treatment of heart failure as tolerated. Presentation with fulminant disease requires rapid evaluation and therapy as discussed earlier. For patients with subacute presentation, randomized trials have shown no benefit of immunosuppression with glucocorticoid combinations or intravenous immunoglobulin, even when the biopsy is positive for lymphocytic infiltrates; yet, immunosuppression is often used even in the absence of evidence of benefit, in part due to perceived analogy to acute cardiac transplant rejection. Animal models have shown that viral replication and myocardial injury can be worsened by immunosuppression during the early phase of infection; however, patients with persistent inflammatory myocarditis and a progressive downhill course over weeks may be treated empirically with glucocorticoids in an attempt to avoid the need for cardiac transplantation.
The true prognosis of viral myocarditis is not known, as most unrecognized cases probably resolve spontaneously, while others progress to cardiomyopathy without other obvious cause. However, among patients who have truly recent onset cardiomyopathy of less than 3–6 months’ duration without other apparent etiology, almost half will have major improvement in left ventricular ejection fraction during the subsequent 6–12 months. Those patients in whom left ventricular ejection fraction and dimensions return to normal are usually considered to have residual subclinical cardiomyopathy. Neurohormonal antagonist therapy is usually continued indefinitely as tolerated, with dose adjustments to avoid side effects.
Specific viruses
In humans, viruses are often suspected but rarely confirmed as the direct cause of myocarditis. Often implicated is the picornavirus family of RNA viruses, with the enteroviruses Coxsackie, echovirus, and poliovirus. Influenza, another RNA virus, is implicated in myocarditis with varying frequency from year to year as the epitopes change. Of the DNA viruses, adenovirus, variola (smallpox) and vaccinia (smallpox vaccine), and the herpesviruses (Varicella zoster, Cytomegalovirus, and Epstein-Barr virus) are well recognized as causes of myocarditis. From genetic analyses of biopsy tissue, parvovirus B19, coxsackie, adenovirus, and Epstein-Barr virus are the agents most often implicated. The role of parvovirus B19 as a cause of myocarditis or cardiomyopathy is difficult to determine, as almost half of individuals show evidence of prior infection with this small DNA virus that causes “fifth disease” in children.
Human immunodeficiency virus (HIV) has been associated with echocardiographic abnormalities in 10–40% patients with clinical disease. Cardiomyopathy in HIV may result from cardiac involvement with other associated viruses, such as cytomegalovirus and hepatitis C. Antiviral drugs to treat chronic HIV can cause cardiomyopathy, both directly as cardiotoxins and through drug hypersensitivity. The clinical picture may be complicated by pericardial effusions and pulmonary hypertension. There is a high frequency of lymphocytic myocarditis found at autopsy, and viral particles have been demonstrated in the myocardium in some cases, consistent with direct causation.
Hepatitis C has been repeatedly implicated in cardiomyopathy, particularly in Germany and Asia. Cardiac function may improve after interferon therapy. As this cytokine itself often depresses cardiac function transiently, careful coordination of administration and ongoing clinical evaluation are critical. Involvement of the heart with hepatitis B is uncommon but can be seen when associated with systemic vasculitis (polyarteritis nodosa).
Other viral infections in which cardiac involvement is specifically implicated, beyond the depression of cardiac function during any systemic cytokine activation, include mumps, respiratory syncytial virus, the arboviruses (dengue fever and yellow fever), and arenaviruses (Lassa fever).
Parasitic myocarditis
Chagas’ disease is the third most common parasitic infection in the world and the most common cause of cardiomyopathy. The protozoan Trypanosoma cruzi (T. cruzi) is usually transmitted by the bite of the reduviid bug, endemic in the rural areas of South and Central America. Transmission can also occur through blood transfusion, organ donation, from mother to fetus, and occasionally orally. While programs to eradicate the insect vector have decreased the prevalence from about 16 million to less than 10 million in South America, cases are increasingly recognized in Western developed countries. Approximately 100,000 affected individuals are currently living in the United States, most of whom contracted the disease in endemic areas.
The acute phase of Chagas’ disease with parasitemia is usually unrecognized, but in fewer than 5% of cases presents clinically within a few weeks of infection, with nonspecific symptoms or occasionally with acute myocarditis and meningoencephalitis. In the absence of anti-parasitic therapy, the silent stage progresses slowly over 10–30 years in almost half of patients to manifest in the cardiac and gastrointestinal systems in the chronic stages. Survival is less than 30% at 5 years after the onset of overt clinical heart failure.
Multiple pathogenetic mechanisms are implicated. The parasite itself can cause myocyte lysis and primary neuronal damage, and specific immune responses may recognize the parasites or related antigens and lead to chronic immune activation in the absence of detectable parasites. Molecular techniques have revealed persistent parasite DNA fragments in infected individuals. Further evidence for persistent infection is the eruption of parasitic skin lesions during immunosuppression after cardiac transplantation. As in postviral myocarditis, the relative roles of persistent infection and of secondary autoimmune injury have not been resolved (Fig. 21-5). An additional factor in progression of Chagas’ disease is the autonomic dysfunction and microvascular damage that may contribute to cardiac and gastrointestinal disease.
Features typical of Chagas’ disease are conduction system abnormalities, particularly sinus node and atrioventricular (AV) node dysfunction and right bundle branch block. Atrial fibrillation and ventricular tachyarrhythmias also occur. Small ventricular aneurysms are common, particularly at the apex. The dilated ventricles are particularly thrombogenic, giving rise to pulmonary and systemic emboli. The serologic enzyme-linked immunosorbent assay (ELISA) for the IgM has largely replaced the previous complement fixation test for diagnosis.
Treatment of the advanced stages focused on the clinical manifestations of the disease, with heart failure regimens, pacemaker-defibrillators, and anticoagulation; however, increasing emphasis is placed on antiparasitic therapy even in chronic disease. The most common effective anti-parasitic therapies are benznidazole and nifurtimox, both associated with multiple severe reactions, including dermatitis, gastrointestinal distress, and neuropathy. Patients without major extracardiac disease have occasionally undergone transplantation, after which they require lifelong therapy to suppress reactivation of infection.
African trypanosomiasis infection results from the tsetse fly bite and can occur in travelers exposed during trips to Africa. The West African form is caused by Trypanosoma brucei gambiense and progresses silently over years. The East African form caused by T. brucei rhodesiense can progress rapidly through perivascular infiltration to myocarditis and heart failure, with frequent arrhythmias. The diagnosis is made by identification of trypanosomes in blood, lymph nodes, or other affected sites. Development of optimal drug regimens remains limited, and depends on the type and the stage (hemolymphatic or neurologic).
Toxoplasmosis is contracted through undercooked infected beef or pork, transmission from feline feces, organ transplantation, transfusion, or maternal-fetal transmission. Immunocompromised hosts are at greatest risk for reactivation of latent infection from cysts. The cysts have been found in up to 40% of autopsies of patients dying from HIV infection. Toxoplasmosis may present with encephalitis or chorioretinitis, and in the heart can cause myocarditis, pericardial effusion, constrictive pericarditis, and heart failure. The diagnosis may be suspected in an immunocompromised patient with myocarditis and serologic evidence of toxoplasmosis. Fortuitous sampling may reveal the cysts in the myocardium. Combination therapy can include pyrimethamine and sulfadiazine or clindamycin.
Trichinellosis is caused by Trichinella spiralis larva ingested with undercooked meat. Larva migrating into skeletal muscles cause myalgias, weakness, and fever. Periorbital and facial edema, and conjunctival and retinal hemorrhage may also be seen. Although the larva may occasionally invade the myocardium, clinical heart failure is rare, and when observed, attributed to the eosinophilic inflammatory response. The diagnosis is made from the specific serum antibody and is further supported by the presence of eosinophilia. Treatment includes antihelminthic drugs and glucocorticoids if inflammation is severe.
Cardiac involvement with Echinococcus is rare, but cysts can form and rupture in the myocardium and pericardium.
Bacterial infections
Most bacterial infections can involve the heart occasionally through direct invasion and abscess formation, but do so rarely. More commonly, contractility is depressed globally in severe infection and sepsis through systemic inflammatory responses. Diphtheria specifically affects the heart in almost one-half of cases and is the most common cause of death in patients with this infection. Once a disease of children, the prevalence of vaccines has shifted the incidence of this disease to countries where immunization is not routine and to older populations who have lost their immunity. The bacillus releases a toxin that impairs protein synthesis and may particularly affect the conduction system. The specific antitoxin should be administered as soon as possible, with higher priority than antibiotic therapy. Other systemic bacterial infections that can involve the heart include brucellosis, chlamydophila, legionella, meningococcus, mycoplasma, psittacosis, and salmonellosis, for which treatment is directed at the systemic infection.
Clostridial infections cause myocardial damage from the released toxin. Gas bubbles can be detected in the myocardium, and occasionally abscesses can form in the myocardium and pericardium. Streptococcal infection with β-hemolytic streptococci is most commonly associated with acute rheumatic fever, and is characterized by inflammation and fibrosis of cardiac valves and systemic connective tissue, but can also lead to a myocarditis with focal or diffuse infiltrates of mononuclear cells.
Tuberculosis can involve the myocardium directly as well as through tuberculous pericarditis, but rarely does so when the disease is treated with antibiotics. Whipple’s disease is caused by Tropheryma whippleii. The usual manifestations are in the gastrointestinal tract, but pericarditis, coronary arteritis, valvular lesions, and occasionally clinical heart failure may also occur. Multidrug antituberculous regimens are effective, but the disease tends to relapse even with appropriate treatment.
Other infections
Spirochetal myocarditis has been diagnosed from myocardial biopsies containing Borrelia burgdorferi that causes Lyme disease. Lyme carditis most often presents with arthritis and conduction system disease that resolves within 1–2 weeks of antibiotic treatment, only rarely causing clinical heart failure.
Fungal myocarditis can occur due to hematogenous or direct spread of infection from other sites, as has been described for aspergillosis, actinomycosis, blastomycosis, candidiasis, coccidioidomycosis, cryptococcosis, histoplasmosis, and mucormycosis. However, cardiac infection is rarely the dominant clinical feature of these infections.
The rickettsial infections, Q fever, Rocky Mountain spotted fever, and scrub typhus, are frequently accompanied by ECG changes, but most clinical manifestations relate to systemic vascular involvement.
NONINFECTIVE MYOCARDITIS
Myocardial inflammation can occur without apparent preceding infection. The paradigm of noninfectious inflammation without infection is cardiac transplant rejection, from which we have learned that myocardial depression can develop and reverse quickly, that non-cellular mediators such as antibodies and cytokines play a major role in addition to lymphocytes, and that myocardial antigens are exposed by prior physical injury and viral infection.
The most commonly diagnosed noninfectious inflammation is granulomatous myocarditis, including both sarcoidosis and giant cell myocarditis. Sarcoidosis is a multisystem disease most commonly affecting the lungs, presenting in young adults with higher prevalence in African-American males. Patients with pulmonary sarcoid are at high risk for cardiac involvement, but cardiac sarcoidosis may also occur without clinical lung disease in middle-aged whites of both genders. Regional clustering of the disease supports the suspicion that the granulomatous reaction is triggered by an infectious or environmental allergen not yet identified.
The sites and density of cardiac granulomata, the time course, and the degree of extracardiac involvement are remarkably variable. Patients may present with rapid onset heart failure and ventricular tachyarrhythmias, conduction block, chest pain syndromes, or minor cardiac findings in the setting of ocular involvement, an infiltrative skin rash, or a nonspecific febrile illness. They may also present less acutely after months to years of fluctuating cardiac symptoms. When ventricular tachycardia or conduction block dominate the initial presentation of heart failure without coronary artery disease, suspicion should be high for these granulomatous myocarditides.
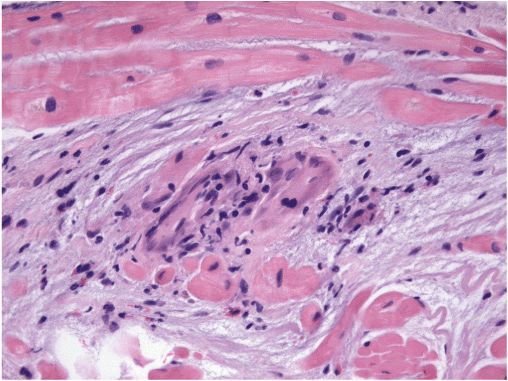
Depending on the time course, the ventricles may appear restrictive or dilated, at times with right ventricular predominance. Small ventricular aneurysms are common. Computed tomography of the chest often reveals pulmonary lymphadenopathy even in the absence of clinical lung disease. Metabolic imaging (positron emission tomography [PET]) of the whole chest can highlight active sarcoid lesions that are avid for glucose. Magnetic resonance imaging (MRI) of the heart can identify areas likely to be inflammatory. To rule out chronic granulomatous infections, the diagnosis usually requires pathologic confirmation. Biopsy of enlarged mediastinal nodes may provide the highest yield. The scattered granulomata of sarcoidosis can be missed on cardiac biopsy (Fig. 21-7).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


