Chapter 18 Ventricular Septal Defect with Pulmonary Stenosis
Etienne-Louis Arthur Fallot’s classic publication, L’Anatomie Pathologique de La Maladie Bleue (Figure 18-1A), appeared in 1888 in an obscure journal published in Marseille where Fallot lived throughout his life.1 The malformation reported by Fallot was originally described 1671 by Niels Stensen,2 better known by his Latinized name, Nicholas Steno, who was equally distinguished as anatomist, geologist, and theologian.3,4 Steno wrote, “When I opened the right ventricle … the probe that was passed forward and upward along the interventricular septum entered directly into the aorta just as readily as the probe passed from left ventricle into aorta. The same aortic canal … was common to both ventricles. Thus, the aorta receives blood from both ventricles at the same time … as it partly straddles the right ventricle”4 (Figure 18-1B). In 1872, 16 years before Fallot’s publication, Sir Thomas Watson5 wrote, “The septum between the ventricles was imperfect in its upper part; and the aorta belonged as much to one ventricle as to the other. The pulmonary artery would not admit a goose-quill; the walls of the right ventricle were as thick as those of the left.” The anatomic and clinical features of Fallot’s tetralogy were also described by Eduard Sandifort (1777),4,6 William Hunter (1784),7 James Hope (1839),8 and Thomas Peacock (1866).9 Fallot made an anatomic diagnosis at the bedside, was proven right at postmortem, and coined the term tetralogy.10 He said, “This malformation consists of a true anatomo-pathologic type represented by the following tetralogy: 1) stenosis of the pulmonary artery; 2) interventricular communication; 3) deviation of the origin of the aorta to the right; 4) hypertrophy, almost always concentric, of the right ventricle.”10 Fallot requested that no eulogy be published after his death, but the tetralogy that bears his name remains one of the most familiar eponyms in cardiovascular medicine. The incidence rate is estimated at 1 in 3600 live births.11,12
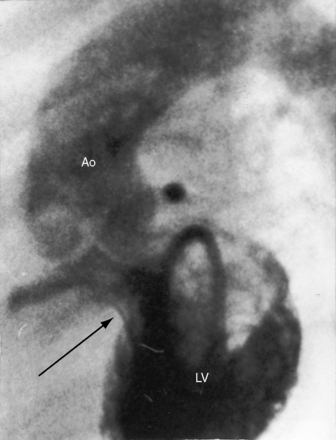
The four salient anatomic components of Fallot’s tetralogy result from a specific morphogenetic abnormality: malalignment of the infundibular septum.13 In the normal heart, division of the fetal conotruncus culminates in alignment of the infundibular septum with the muscular trabecular septum. In Fallot’s tetralogy, the infundibular septum deviates anteriorly and cephalad and is therefore not aligned with the trabecular septum, creating a ventricular septal defect at the site of malalignment. The deviation of the infundibular septum encroaches on the right ventricular outflow tract and causes infundibular stenosis and a biventricular (overriding) aorta (Figure 18-2).13 The degree of override and the size of the biventricular aorta are determined chiefly by the degree of malalignment, but aortic size is also influenced by an inherent medial abnormality.14 The nonrestrictive malaligned ventricular septal defect accounts for systemic systolic pressure in the right ventricle and concentric right ventricular hypertrophy.
Malaligned ventricular septal defects are located in the perimembranous septum with extension into the infundibular septum.13 Atrioventricular conduction is normal. The crest of the muscular trabecular septum forms the floor of the defect, which is roofed by the valve of the overriding aorta, setting the stage for aortic regurgitation. Subarterial ventricular septal defects, which are more frequent among Asians, are understandably accompanied by aortic regurgitation because a supporting infundibulum is absent.15 Rarely, the ventricular septal defect is part of an atrioventricular septal defect (see Chapter 15).16–18 Muscular ventricular septal defects sometimes coexist but usually close spontaneously in the first year of life. Occasionally, a nonrestrictive malaligned defect is reduced in size by intrusion of accessory or excessive tricuspid valve tissue (see Figure 18-18) that is fixed to the edges of the defect by short chordae tendineae or tethered by long chordae, which permit wide excursions through the defect.19
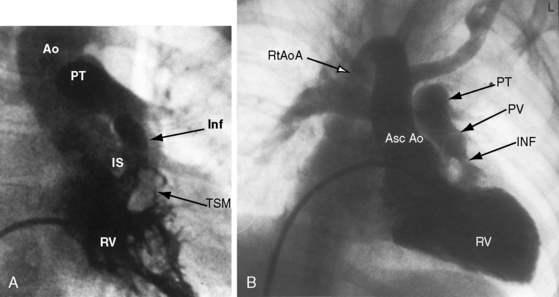
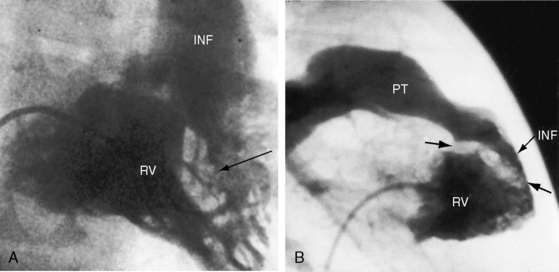
Malalignment of the infundibular septum is the essential but not the only cause of obstruction to right ventricular outflow,13 which also results from hypertrophy of the septoparietal trabeculations, the trabecula septomarginalis, and the infundibular septum (Figure 18-3A). Anterior and cephalad malalignment of the infundibular septum can narrow the entire right ventricular outflow tract (see Figure 18-3).13,20 The pulmonary valve is frequently stenotic and bicuspid (see Figure 18-3B),21 and less frequently unicommissural unicuspid.22 Occasionally, the main site of obstruction is a hypoplastic pulmonary annulus or the ostium of the infundibulum (Figure 18-4).23 The pulmonary trunk, its bifurcation, and its right and left branches may be segmentally or diffusely hypoplastic (Figure 18-5B).23 Rarely, the pulmonary arteries cross as they proceed to their respective lungs.24 Even more rarely, the tetralogy is associated with the scimitar syndrome.25
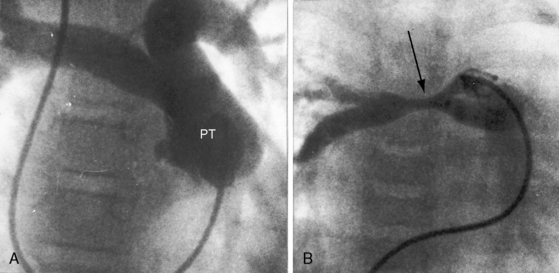
Pulmonary atresia with Fallot’s tetralogy is the ultimate expression of severity.26 The right ventricle terminates blindly against an atretic pulmonary valve or against imperforate muscle (Figures 18-6A and 18-7B). The pulmonary trunk is either a vestigial cord or a hypoplastic funnel-shaped channel that widens as it approaches the bifurcation. The proximal pulmonary arteries are hypoplastic (Figure 18-7D) and may be discontinuous.26 The entire right ventricular output enters the systemic circulation via the nonrestrictive malaligned ventricular septal defect (see Figures 18-6A and 18-7B). The biventricular aorta is dilated (see Figures 18-6A and 18-7A, B) and often continues as a right aortic arch (Figure 18-8A). The lungs are perfused by systemic-to-pulmonary arterial collaterals (see Figures 18-6B and 18-7C, D), on which survival depends (see subsequent).27,28 Exceptionally, the pulmonary circulation is supplied primarily, if not exclusively, by a long, narrow sigmoid-shaped ductus arteriosus (see Figure 18-38B) that is structurally a muscular systemic artery similar to a systemic arterial collateral. This ductal structure is appropriate for intrauterine flow, which is directed from the aorta into the pulmonary artery.
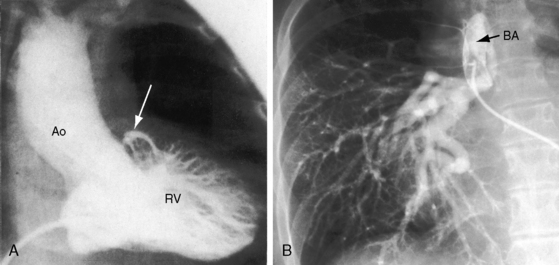
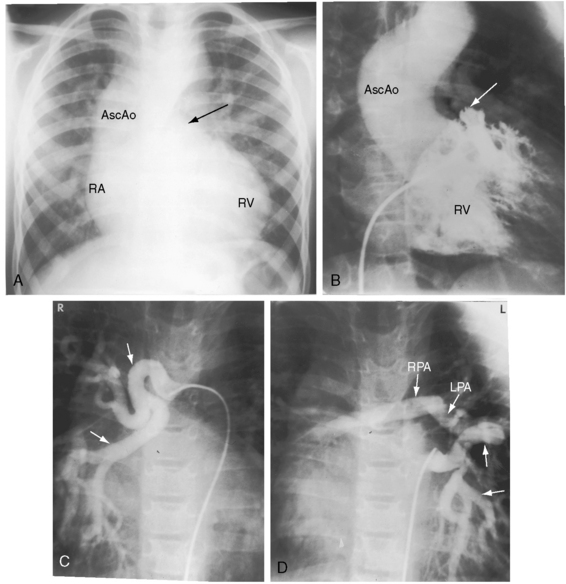
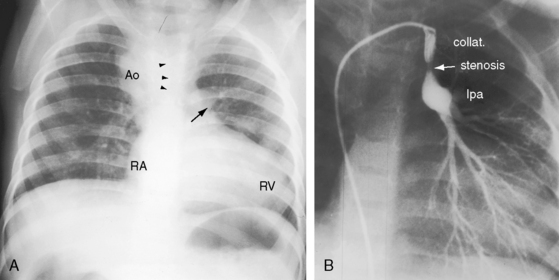
One of the most characteristic features of Fallot’s tetralogy with pulmonary atresia is a pulmonary circulation supplied entirely by collateral arteries that serve both a nutritive function and a respiratory function (gas exchange). The three types of arterial blood supply to the lungs include systemic arterial collaterals; the distinctive ductus arteriosus, which is a muscular systemic artery (see previous); and small diffuse pleural arterial plexuses.27Systemic arterial collaterals are classified according to their origins as: (1) bronchial, which originate where their name indicates and anastomose to pulmonary arteries within the lung (see Figure 18-6B)28,29; (2) direct systemic arterial collaterals, which originate from the descending aorta, enter the hilum, and then assume the structure and distribution of intrapulmonary arteries (see Figure 18-7C, D)28; and (3) indirect systemic arterial collaterals, which originate from the internal mammary, innominate, and subclavian arteries and anastomose to proximal pulmonary arteries outside the lung (see Figure 18-8B).28 Bronchial arterial collaterals are characterized by intrapulmonary anastomoses, direct arterial collaterals by hilar anatomoses, and indirect arterial collaterals by extrapulmonary anatomoses.28 Thus, systemic arterial collaterals anatomose with pulmonary arteries in three locations: (1) intrapulmonary; (2) extrapulmonary; and (3) hilar.28 All three major types of collaterals are present when Fallot’s tetralogy occurs with pulmonary atresia, but only bronchial collaterals are present when the tetralogy occurs with pulmonary stenosis, irrespective of severity.28 About 10% of arterial collaterals originate from coronary arteries.30–32 The pulmonary circulation is effective in gas exchange regardless of the type of systemic arterial collateral.
Direct aortic to pulmonary collaterals originate from intersegmental branches of the dorsal aorta during the third and fourth weeks of gestation.28,29Bronchial arterial collaterals develop in the ninth gestational week after the paired intersegmental arteries have been resorbed28,29 and do not coexist with direct aortic collaterals.28Indirect collaterals arise later in gestation and therefore coexist with bronchial collaterals but not with direct aortic collaterals.28 A particular collateral artery supplies a particular segment of lung, but duplicate blood supplies occasionally occur. A single type of collateral usually predominates in a given patient.28 Lung growth and survival depend on the size and patency of the collateral arteries. Diminished pulmonary blood flow adversely effects the growth of peripheral pulmonary arteries.28
Systemic arterial collaterals have a strong tendency to harbor intimal cushions (proliferations) that serve as sites of potential segmental stenosis (see Figure 18-8B).28 In the absence of these obstructing cushions, large collateral arteries transmit systemic arterial pressure into the pulmonary vascular bed, resulting in morphologic changes analogous to pulmonary vascular disease (see Chapter 14).28,33 Although stenotic sites protect the intrapulmonary resistance vessels from systemic pressure, regional pulmonary blood flow is compromised.
In 1947, Taussig observed that the ductus arteriosus was not structurally normal in Fallot’s tetralogy with pulmonary atresia.34,35 The normal fetal ductus functions as a conduit for right ventricular flow into pulmonary trunk, a function that cannot be served when pulmonary atresia diverts the entire right ventricular output into the aorta via the nonrestrictive malaligned ventricular septal defect. Not surprisingly, the ductus is malformed or absent when pulmonary atresia exists from early fetal life. Absence of a ductus indicates that normal intrauterine ductal function was usurped, rendering the ductus superfluous. If a ductus is present, it is represented by a long narrow branch of the aorta that carries systemic arterial blood from the aorta into the pulmonary trunk.27 This ductus is narrow because it delivers blood only to the lungs, which represents no more than 5% to 10% of the combined ventricular output. The ductus is long because it first runs distally, diverging from the aortic arch, and then turns back to join the proximal left pulmonary artery.
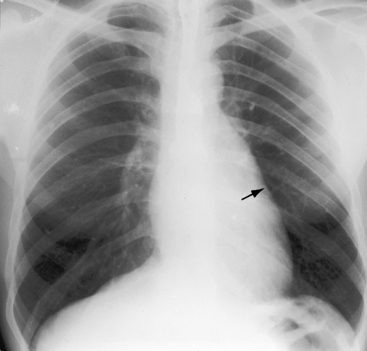
Aortic regurgitation in Fallot’s tetralogy occurs because the malaligned ventricular septal defect is partially roofed by the aortic valve.13,36 Herniation of aortic cusps is more frequent with an isolated subarterial ventricular septal defects than with Fallot’s tetralogy, a difference ascribed to dissimilar flow patterns and their impact on the aortic valve. In cyanotic Fallot’s tetralogy, the aortic valve is not subjected to turbulent flow because the left ventricle ejects directly into the aorta without generating a left-to-right jet. Nevertheless, there is an age-related increase in aortic regurgitation,37 in part as a result of progressive aortic root dilation associated with an inherent medial abnormality.14,38,39 Aortic regurgitation causes volume overload of both ventricles because the aorta is biventricular.37 The incompetent aortic valve is susceptible to infective endocarditis, which can suddenly and catastrophically augment the degree of biventricular regurgitation (see section The History).37 A right aortic arch is a feature of Fallot’s tetralogy.40 Its incidence rate increases as the severity of right ventricular outflow obstruction increases (see Figure 18-3) and reaches approximately 25% with pulmonary atresia (see Figures 18-6A and 18-8A).
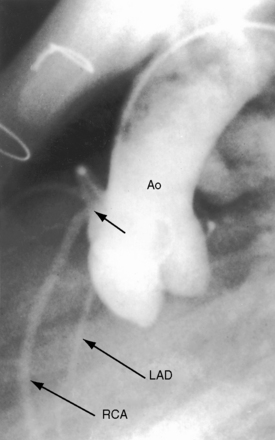
Anomalous origin and distribution of coronary arteries are common (see Chapter 32)30,41,42 and may be of no functional importance but are of considerable surgical importance. The incidence of coronary artery anomalies is influenced by aortopulmonary rotation43 and is higher when the aortic root is anterior to or side-by-side the pulmonary trunk. The most common anomalies are origin of a conus artery or the left anterior descending artery from the right coronary artery or from the right sinus of Valsalva (Figure 18-9).41 Origin of a single coronary artery from the right sinus of Valsalva is less common. Relatively frequent are fistulous communications between coronary arteries and the pulmonary artery30,44 or the right atrium and between coronary arteries and bronchial arteries.32 Rarely, the left anterior descending coronary artery originates from the pulmonary artery,45 or the left coronary artery is intramural.46
Etienne-Louis Fallot recognized that “… at times, there is an additional entirely accessory defect, namely, patency of the foramen ovale.”10 An atrial septal defect occasionally coexists with Fallot’s tetralogy, but the term pentalogy is longer used. Rarely, the tetralogy is associated with total anomalous pulmonary venous connection and an atrial septal defect.47
The combination of right ventricular outflow obstruction and ventricular septal defect is not confined to Fallot’s tetralogy. Pulmonary valve stenosis occasionally occurs with an isolated perimembranous ventricular septal defect (Figure 18-10). The degree of stenosis varies from trivial to severe, the size of the ventricular septal defect varies from small to nonrestrictive, and right ventricular systolic pressure varies from normal to suprasystemic. Other examples of the combination include pulmonary valve stenosis with a muscular ventricular septal defect,48 obstruction to right ventricular outflow caused by protrusion of a large ventricular septal aneurysm into the right ventricular outflow tract (Figure 18-11), and double-chambered right ventricle with a perimembranous49–51 or a malaligned ventricular septal defect (Figure 18-12).20,52 Sir Arthur Keith in his 1909 Hunterian lecture focused on obstructing right ventricular muscular bundles, which consist of an hypertrophied moderator band, an hypertrophied trabecula, or a fibromuscular diaphragm,53 with obstruction ranging from nil to severe to complete.50,54 A ventricular septal defect that communicates with the proximal high-pressure compartment results in a right-to-left shunt.50
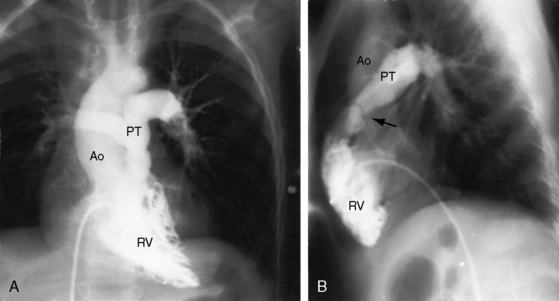
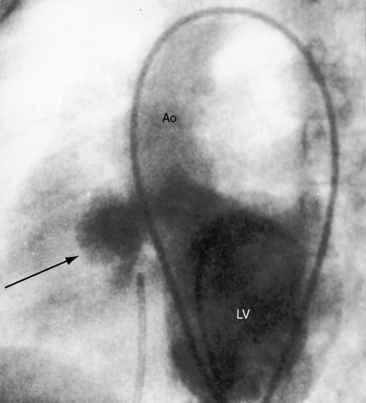
Taussig55 described two infants with a loud holosystolic murmur at the lower left sternal border and increased pulmonary blood flow, prompting the diagnosis of ventricular septal defect. Several years later, both patients were cyanotic with pulmonary stenotic murmurs and decreased pulmonary blood flow, appropriate for the diagnosis to Fallot’s tetralogy. Gasul formalized the notion that progressive infundibular obstruction sometimes occurs with ventricular septal defect (Figure 18-13), and confirmatory reports soon appeared.56–58 The acquired obstruction usually results from hypertrophy of right ventricular muscle bundles, and only rarely from a malaligned infundibular septum.57
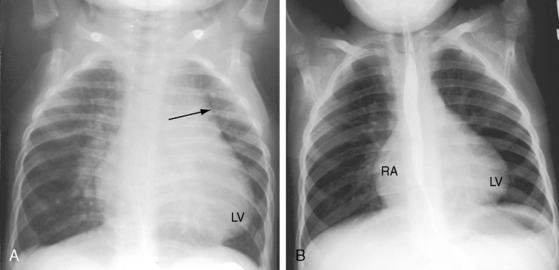
The physiologic consequences of Fallot’s tetralogy depend essentially on two variables: the degree of obstruction to right ventricular outflow and, to a lesser extent, systemic vascular resistance. The magnitude and direction of the shunt are determined by the resistance at the site of pulmonary stenosis relative to systemic vascular resistance. When pulmonary stenosis offers lesser resistance, the shunt is left-to-right. When the resistances are equal, the shunt is balanced. When right ventricular outflow resistance exceeds systemic resistance, the shunt is right-to-left. The amount of aortic override is not the issue, although the degree of override tends to coincide with the degree of right ventricular outflow obstruction, which is the issue. When right ventricular blood preferentially flows into the aorta, pulmonary blood flow falls reciprocally, so the left side of the heart is underfilled.59,60 The ultimate expression of right ventricular outflow obstruction is pulmonary atresia, which commits the entire right ventricular output to the aorta. Pulmonary blood flow then depends on systemic arterial collaterals (see previous), which provide the lungs with normal or increased flow, so cyanosis can be mild or even absent.61 Unobstructed flow through arterial collaterals sets the stage for pulmonary vascular disease. Stenoses at intimal cushions in arterial collaterals (see previous) protect the pulmonary vascular bed, but at the price of reduced pulmonary blood flow.
Irrespective of the degree of right ventricular outflow obstruction, right ventricular systolic pressure cannot exceed systemic because the ventricular septal defect is nonrestrictive. Accordingly, a systemic ceiling is placed on the pressure overload that pulmonary stenosis can impose on the right ventricle. When pulmonary stenosis is severe, right ventricular pressure overload is determined by systemic vascular resistance. Increased resistance associated with systemic hypertension or, less commonly, with acquired calcific aortic stenosis (see Figure 18-33B) improves pulmonary blood flow but increases right ventricular afterload. Aortic regurgitation imposes volume load on the already pressure-overloaded right ventricle.
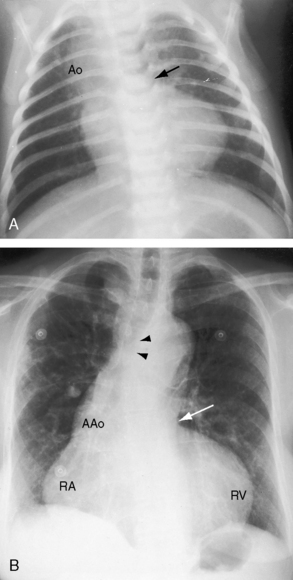
In addition to concentric hypertrophy of the right ventricle, certain morphologic changes are secondary to the physiologic derangements of Fallot’s tetralogy. Tricuspid leaflets develop fibrous thickening because right ventricular systolic pressure is systemic, but the thickened leaflets are seldom incompetent. The right ventricle ejects against systemic resistance without an increase in filling pressure, so right atrial pressure remains normal, and overall systolic function of the hypertrophied right ventricle remains normal (Figure 18-14B, C; see subsequent).60 The underfilled left ventricle tends to be reduced in size with reduced stroke volume.59,60 Low pressure and low flow in the pulmonary circulation alter the small muscular arteries and arterioles and cause thinning of the media with interruption of elastic tissue and widespread thromboses.62–64
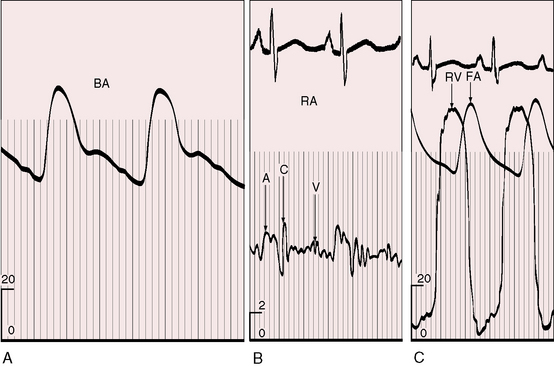
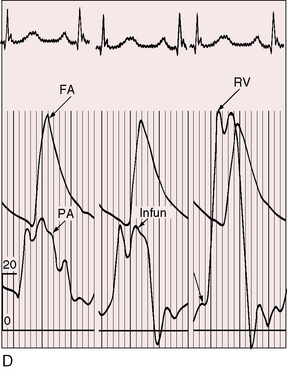
When severe pulmonary stenosis occurs with a restrictive ventricular septal defect, right ventricular systolic pressure exceeds systemic and the hypertrophied right ventricle dilates and fails. A physiologically analogous state exists when accessory tricuspid leaflet tissue partially occludes the malaligned ventricular septal defect (see Figure 18-18).19
History
Gender distribution in Fallot’s tetralogy is approximately equal. The malformation recurs in families and has been reported in siblings,65–67 including triplets,68 and in parents and offspring.67,69 Two brothers with DiGeorge syndrome had Fallot’s tetralogy. Birth weight tends to be lower than normal, and growth and development are generally retarded. Hereditary cardiovascular defects in Keeshond dogs include typical Fallot’s tetralogy and isolated ventricular septal defect with pulmonary stenosis.70 Familial tetralogy has been associated with mutation of the jagged 1 gene,71 with NKX2.5 mutations,72 and with chromosome 22q11.2 deletion.73
The clinical course in early infancy is often benign. Mild to moderate neonatal cyanosis tends to increase, but cyanosis may be delayed for months and is coupled with increased oxygen requirements of the growing infant rather than with progressive obstruction to right ventricular outflow.74 Patients seldom remain acyanotic after the first few years of life, and by 5 to 8 years of age, most children are conspicuously cyanotic, with cyanosis closely coupled to the severity of pulmonary stenosis. Infants with Fallot’s tetralogy and pulmonary atresia are mildly cyanotic or acyanotic when collateral flow is abundant.61
In an analysis of survival patterns based on 566 necropsy cases of Fallot’s tetralogy, two thirds of patients reached their first birthday, approximately half reached age 3 years, and approximately a quarter completed the first decade of life.75,76 The attrition rate was then 6.4% per year with 11% alive at age 20 years, 6% at age 30 years, and 3% at age 40 years.75,76 Nevertheless, Fallot’s tetralogy remains the most common cyanotic congenital heart disease after 4 years of age and constitutes a large proportion of adults with cyanotic congenital heart disease. Fallot recognized this tendency when he wrote, “We have seen from our observations that cyanosis, especially in the adult, is the result of a small number of cardiac malformations well determined. One of these cardiac malformations is much more frequent than others…,” namely, the tetralogy to which he referred.10 Fallot’s oldest patient was 36 years of age.10 Survivals between the fifth and seventh decades are uncommon but not rare.12,77–79 A 64-year-old woman with the tetralogy was diagnosed in 1895 by G.A. Gibson, best known for his description of the continuous murmur of patent ductus arteriosus. In 1929, White and Sprague80 published an account of the American composer Henry F. Gilbert who lived a productive life to age 60 years. Another patient played cricket and football as a schoolboy and survived to age 62 years. Patients without repair have lived to age 75 years,81 78 years,82 84 years,83 and 86 years.84 In Fallot’s tetralogy with pulmonary atresia, life expectancy without surgery is as low as 50% in 1 year and 8% in 10 years,75 but adequate collateral blood flow occasionally permits survival into adolescence and adulthood.18,37,78,85,86 One such patient lived to age 54 years,78 and another lived to age 55 years, despite acquired calcific aortic stenosis and regurgitation (see Figure 18-33B.)
Pregnancy is poorly tolerated in females who reach childbearing age.87 The gestational fall in systemic vascular resistance increases the right-to-left shunt, and labile systemic vascular resistance during labor and delivery results in abrupt oscillations in hypoxemia. Fetal wastage is high,23 and live born infants are dysmature.
The neonatal right ventricle is well equipped to eject against systemic vascular resistance because the nonrestrictive ventricular septal defect permits decompression into the aorta.60 The right ventricle has been analyzed in terms of its inlet, apical trabecular, and outlet portions.88 The apical trabecular portion took up the greatest portion of the overload, the outlet portion had decreased ejecting force, and the inlet portion was not significantly affected.88 Right ventricular failure is uncommon.89 However, biventricular failure in the first few weeks of life accompanies pulmonary atresia with excessive flow through large systemic arterial collaterals.61 Accessory tricuspid leaflet tissue that partially occludes the ventricular septal defect results in suprasystemic right ventricular systolic pressure and the right ventricular failure.19,89 Absence of the pulmonary valve (see subsequent section) results in volume overload of the pressure-overloaded right ventricle.90 Systemic hypertension increases left and right ventricular afterload and can induce right ventricular or biventricular failure.89,91 Acquired calcific stenosis of the biventricular aortic valve imposes increased afterload on both the right and the left ventricles (see Figure 18-33B). Regurgitation of a biventricular aortic valve sets the stage for right ventricular failure by imposing volume overload on the already pressure-overloaded right ventricle.37 Infective endocarditis on an incompetent aortic valve can result in catastrophic acute severe biventricular aortic regurgitation.
Isotonic exercise is accompanied by a fall in systemic vascular resistance in the face of fixed obstruction to right ventricular outflow, increasing venoarterial mixing and significantly influencing the dynamics of O2 uptake and ventilation.92,93 Exercise-induced hypoxemia and increased carbon dioxide content stimulate the respiratory center and the carotid body, provoking hyperventilation that is subjectively perceived as dyspnea.92,93
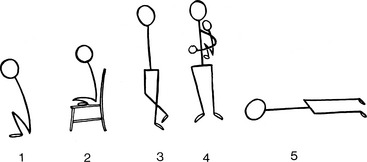
Hypoxic spells, variously called paroxysmal hyperpnea, syncopal attacks, hypoxic or hypercyanotic spells, are dramatic and alarming features of Fallot’s tetralogy.94–96 A typical spell begins with a progressive increase in the rate and depth of breathing and culminates in paroxysmal hyperpnea, deepening cyanosis, limpness, syncope, and occasionally convulsions, cerebrovascular accidents, and death.95,96 Electroencephalographic abnormalities during an hypoxic spell are similar to those of hypoxic episodes of other causes.97 Peak incidence is between the second and sixth month of life, with an occasional spell as early as the first month but comparatively few spells after age 2 years, and only rarely in adults.98 Spells in infants are typically initiated by the stress of feeding, crying, or a bowel movement, particularly after awakening from a long deep sleep.94,95 However, attacks sometimes occur without an apparent precipitating cause, especially in deeply cyanotic infants, although spells are not necessarily related to the degree of cyanosis.95 Spells were originally attributed to infundibular contraction caused by sympathetic stimulation, which was believed to divert right ventricular blood into the aorta,96 but occurrence in patients with pulmonary atresia argued against this theory. It is now believed that vulnerable respiratory control mechanisms, which are especially sensitive after prolonged deep sleep, react to the sudden increase in cardiac output provoked by feeding, crying, or straining, by initiating the following vicious cycle.94,95 As heart rate and cardiac output increase, venous return increases in the face of fixed obstruction to right ventricular outflow, so the right-to-left shunt increases. Infundibular contraction reinforces this pattern but does not initiate it. The increased right-to-left shunt causes a fall in systemic arterial pO2 and pH and a rise in pCO2, a blood gas composition to which a sleep-sensitive respiratory center and carotid body overreact, provoking hyperpnoea, which in turn further increases the cardiac output and perpetuates the cycle. Supraventricular tachycardia and rapid atrial pacing initiate spells by inducing infundibular narrowing, which increases the right-to-left shunt.99 Five mechanisms are therefore involved in the pathogenesis of Fallot spells: (1) an acceleration in heart rate; (2) an increase in cardiac output and venous return; (3) an increase in right-to-left shunt; (4) vulnerable respiratory control centers; and (5) infundibular contraction. Manual compression of the abdominal aorta can abort a spell by decreasing cardiac output and venous return.100 Squatting for relief of dyspnea is a time-honored hallmark of Fallot’stetralogy (Figure 18-15).101,102 In 1784, William Hunter7 made the following observations on the effects of posture: “Any hurry upon his spirits or brisk motion of his body would generally occasion a fit. And for some of the last years of his life he found out by his own observations that when the fit was coming upon him, he would escape it altogether, or at least take considerably from its violence or duration by instantly lying down upon the carpet on his left side, and remaining immovable in that position for about 10 minutes. I saw the experiments made with success.”

Figure 18-15 Typical squatting postures assumed effortlessly in two children with Fallot’s tetralogy.
Taussig described the preference for certain postures other than squatting, namely, the knee-chest position, lying down, or sitting with legs drawn underneath (Figure 18-16).103 Parents may hold their breathless infant upright with its legs flexed against its abdomen (see Figure 18-16, panel 4). Young adults cross their legs during quiet standing or sitting, a relatively ineffective variation. Habitual squatters assume the position effortlessly (see Figure 18-15). The mechanisms by which squatting exerts its beneficial effects are as follows.101,102,104,105 (1) Quiet standing after exercise-induced peripheral vasodilation predisposes to orthostatic hypotension and faintness, a tendency that is exaggerated in hypoxemic patients. Squatting counteracts orthostatic hypotension and diminishes or prevents postexertion orthostatic faintness.102,104 (2) Squatting increases systemic vascular resistance, diverts right ventricular blood into the pulmonary circulation, and increases the amount of oxygenated blood entering the left side of the heart.102,104,106 The left ventricle delivers the larger volume of oxygenated blood into the systemic circulation, so systemic arterial pO2 and pH increase and pCO2 decreases, blunting the stimulus to the respiratory center and carotid body and relieving hyperventilatory dyspnea.104 The effect of squatting on systemic venous return is an even more effective means by which hyperventilatory dyspnea is relieved.104 (3) Isotonic leg exercise reduces the oxygen saturation of venous effluent returning to the heart from the lower extremities. Squatting mechanically curtails lower extremity venous return, decreases the volume of unsaturated venous blood delivered to the heart, and increases the oxygen saturation of right ventricular blood. (4) Right ventricular blood shunted into systemic circulation has a higher oxygen content and pH and a lower pCO2 content.104 (5) The higher pO2 and pH and the lower pCO2 reduce the stimulus to the respiratory center and carotid body and reduce the hyperventilatory dyspnea.
Recurrent hypoxic spells sometimes lead to brain damage and mental retardation. Cerebral venous sinus thromboses and small occult thromboses may become manifest after prolonged hypoxic spells. Hypernasal resonance or nasal speech (velopharyngeal insufficiency) may develop after repeated or prolonged spells because nasal resonance is compromised by improper approximation of the velum (soft palate) and the pharyngeal walls, a disturbance that has been ascribed in part to central nervous system damage caused by hypoxic spells.107
Brain abscess and cerebral embolism add to the list of central nervous system complications.108–110 Iron-deficient erythrocytosis in patients less than 4 years of age increases the risk of cerebral venous sinus thrombosis.110 Wheezing and stridor have been attributed to tracheal compression by an enlarged aorta.14,111 A stenotic pulmonary valve112 and an incompetent aortic valve113 are substrates for infective endocarditis.
The physiologic consequences and clinical course of a nonrestrictive ventricular septal defect are favorably influenced by mild to moderate acquired obstruction to right ventricular outflow (see previous57 and Chapter 17). The clinical picture initially resembles an isolated nonrestrictive ventricular septal defect with large left-to-right shunt (see Figure 18-13A.) With the development of right ventricular outflow obstruction, excessive pulmonary blood flow and volume overload of the left ventricle are curtailed,56,58,74 symptoms related to the left-to-right shunt diminish, and physical development improves. Obstruction to right ventricular outflow may progress sufficiently to reverse the shunt, resulting in late onset cyanosis (see Figure 18-13B).
When a restrictive ventricular septal defect is accompanied by severe pulmonary valve stenosis, the clinical picture resembles isolated pulmonary stenosis with intact ventricular septum (see Chapter 11). A restrictive ventricular septal defect with mild pulmonary stenosis is associated with a conspicuous murmur and few or no symptoms but with the risk of infective endocarditis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


