Chapter 12
Venous Pathology
Peter K. Henke
Veins are complex organs, much like arteries, and well suited to their physiologic purpose. Venous diseases are common in the general population and are influenced by genetics, environment, and acquired conditions (Fig. 12-1). Understanding the basic physiologic and molecular responses to venous injury is essential for designing effective and safe therapies.
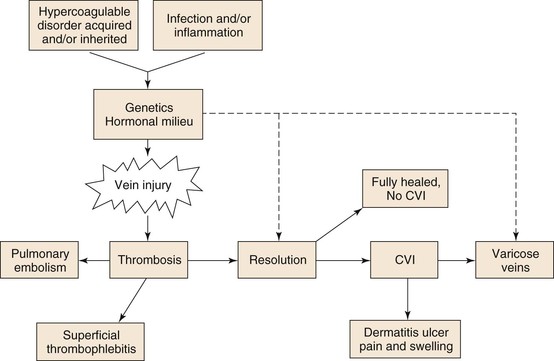
Figure 12-1 Hypercoagulable, acquired, or inherited disorders as well as infection, inflammation, and stasis, in conjunction with the patient’s physiologic milieu and genetics, affect vein wall injury. This injury may lead to thrombosis, vein injury, or varicose veins. Of those patients in whom thrombosis develops, a pulmonary embolism may occur or thrombosis resolves with no sequela. More commonly however, chronic venous insufficiency (CVI) develops, which may potentiate varicosities as well as lead to postthrombotic vein injury and dermatitis with ulcers, pain, or swelling.
Basic Considerations
Venous Cellular and Developmental Concepts
The physiology of veins has become better studied over the last several decades. Veins are not passive conduits but rather have regional and anatomically specific functions that contribute in diverse ways to organism homeostasis. Numerous differences exist between arteries and veins, including the physical structure of the vein wall, the presence of valves, the fact that veins carry deoxygenated blood, and the magnitude of shear and wall stresses (Table 12-1).
Table 12-1
Artery and Vein Comparisons
| Artery | Vein | |
| Embryology | Branchial arches | Cardinal veins |
| Cellular markers | EphB2, delta-like ligand 4, AIK-1, EPAS-1 | EphB4, NRP2, COUP-TF II |
| Hemostasis mechanism | Platelet-mediated, extrinsic pathway, exposed subendothelial collagen | Tissue factor– and fibrin-mediated, extrinsic pathway, cell adhesion molecules, von Willebrand factor –platelet |
| Response to injury | Plaque formation; occlusion | Thrombosis; fibrosis |
| Vasoregulation mechanisms | Adrenergic > endothelin Nitric oxide | Endothelin > adrenergic Nitric oxide |
| Anticoagulant mechanisms | Thrombomodulin, tissue plasminogen activator, tissue factor pathway inhibitor | Thrombomodulin, endothelial protein C receptor , urinary plasminogen activator |
The embryonic vascular primordia of both arteries and veins are progenitor hematopoietic stem cells that arise in the yolk sac. These stem cells, called “blood islands,” separate into peripheral cells that differentiate into formed vascular structures. The initial angiogenic capillaries, which are essentially endothelial tubes, recruit mesenchymal stem cells that become smooth muscle cells, secrete matrix, and are followed later by adventitial fibroblasts.
Research now suggests that vascular morphogenesis is primarily related to morphologic changes in endothelial stem cells that depend on vascular endothelial growth factor (VEGF), the NOTCH signaling pathway, and the transforming growth factorβ (TGF-β)/bone morphogenic protein (BMP) signaling pathways.1 Angiogenic sprouting is dependent on VEGF, whereas arterial and vein identity depends on NOTCH signaling. Not surprisingly, arteries and veins express different embryologic endothelial molecular markers. For example, venous endothelium–specific genes include EPHB4, neurophilin-2, and COUP-TFII.2 These signaling pathways may mediate venous and arterial development. For instance, suppression of NOTCH signaling by COUP-TFII (chicken ovalbumin upstream promoter transcription factor II) is essential for vein identity.3 The pathways of vascular morphogenesis have also become better delineated in zebrafish and other basic models. Disruption of the bone morphogenic protein–chordin pathway results in severe experimental venous malformations in experimental models, depending on where anatomically the inhibition occurs.4
Although embryonic vasculogenesis may differ from adult vasculogenesis, interaction between the endothelium and the basement membrane matrix is critical for certain processes, including endothelial cell migration, proliferation, and survival. Laminin, integrins, and glycoproteins are all important components that direct intracellular signaling.5 How these processes specifically relate to venous development has not been as well studied but is probably similar to the relationship in arteriogenesis.
The macroembryology of veins is well described and is more variable than arterial development. The embryologic origin of the venous system is the cardinal veins, which form the main venous drainage system in the embryo, consisting of the anterior and posterior segments. Other venous structures are formed between the fifth and seventh weeks of embryogenesis, including the supracardial and sacrocardinal veins. Because there are multiple embryologic trunks, numerous anomalies can develop from the pairing of these basic venous structures and contribute to the variability of venous anatomy in individuals.
The histologic structure of veins is similar to that of all vessels, consisting of an intima (the endothelium with basement membrane), a thin medial layer composed of smooth muscle cells and a surrounding matrix, and an adventitia. In normal veins, the muscle layers are composed of smooth muscle cells, which are spindle shaped and have a contractile phenotype.6 These cells lie in close proximity to one another in parallel arrays and are surrounded by bundles of regularly arranged collagen fibers.
The vein wall endothelium separates the intravascular and extravascular spaces and has a regulated and selective function.7 Endothelial gap junctions, although nonfenestrated, regulate the diapedesis of leukocytes as well as efflux of plasma and macromolecules, which has been best studied on the high endothelial venules. Vein wall endothelial cells are shorter and broader than arterial wall cells and are not aligned to flow, in contrast to arterial endothelium.2
Venous Biomechanics
The biomechanics of veins suggest excellent adaptation for their function. Veins allow a very large volume capacitance, and tonal regulation, to rapidly redistribute overall blood volume. Approximately, 60% to 80% of circulating blood is stored in the venules and the systemic veins at any given time. The function of the blood capacitance system, via vasoregulation, is to maintain the filling pressure of the heart as well as to compensate for orthostatic changes. The physiology of venous blood flow in the limb related to the calf muscle pump and other actions is detailed in Chapter 11.
Everyday activities and changes in body position cause large changes in venous pressure. The average venous pressure at the foot is approximately 100 mm Hg in a person 5 feet 10 inches tall and weighing 75 kg. This pressure drops significantly with ambulation and while the person is recumbent. The venous valves are endothelium-lined folds of tunica intima that allow directional flow and contribute to this pressure reduction as well as maintaining prograde blood flow. To accommodate pressure and volume changes, veins undergo complex alterations in shape, depending on the blood volume, resistance, and the amount of blood flow within the system. Less vascular resistance occurs with a circular shape than an elliptical shape, and thus as venous volume increases, resistance to flow lessens.
Unlike arteries, large veins lack an extensive elastic lamella (composed of elastin) but exhibit marked distensibility. Veins have a much smaller ratio of wall thickness to radius and higher incremental distensibility in the low-pressure range then arteries do, thus indicating that the elastic modulus of veins can greatly exceed the stress modulus of arteries. As a result, veins have a high breaking pressure, nearly four atmospheres.8 Much of the stress-bearing function of the vein wall may depend on its smooth muscle cell and elastin content, in contrast to the abundance of collagen in the arterial wall. Indeed, vein wall compliance is decreased after experimental venous thrombosis (VT) injury, which correlates with its increased collagen content,9 and disrupted elastin, as measured histologically.10
Venous Vasoregulation
Whereas the elastic properties of the vein wall provide passive tone, active vasoregulation is provided by smooth muscle cells in the medial layer mediated through sympathetic nerves, as well as vasoactive circulating mediators, as in arteries. Central responses include changes in hormones, body temperature, blood volume, physiologic stress, and other conditions. Typical adrenergic agonists are noradrenalin and epinephrine, whereas acetylcholine causes both constriction and relaxation. Locally, vasodilation and vasoconstriction are mediated by endothelium-derived relaxing factor, namely, nitric oxide (NO) and the constricting agent endothelin-1. Stimulators for NO include muscarinic activation, thrombin, and α2-adrenergic agents. Interestingly, the venous endothelium synthesizes less NO and more endothelin-1 than the endothelium of arteries.11 However, the endothelium is a major regulator of venous tone, through similar mechanisms that regulate arterial tone.12 The vein wall also produces vasoactive prostanoids, both intraluminal and extraluminal, that alter tone. For example, prostacyclin synthesis in the vein wall is stimulated by numerous substances and by mechanical stretching and promotes venodilation. Other direct and indirect vasoregulators include angiotensin II, bradykinin, histamine, serotonin, and vasoactive intestinal peptide.
The local physiologic environment also affects venous tone, particularly hypoxemia and the local pH. For example, decreased pH, elevated PCO2, and elevated lactate values correlate with decreased contractility in isolated vein ring experiments.13 Age-related declines in vasodilatory and vasoconstrictor responses occur and may contribute to the demographics of venous disease, which is much more common with advancing age.14
Blood flow rates can also affect venous tone independently of the pressure. Mechanically, vasoregulation occurs with pulsatile blood flow and increased endothelial shear stress,15 which induces synthesis of various substances. In a manner similar to that in arteries, the Na, K, and Ca pumps are also important at the cellular level of venous regulation. Interestingly, the local ionic environment of vein walls may contribute to development of essential hypertension, with greater amounts of sodium ions in juxtaposition with the vein walls in hypertensive patients than in nonhypertensive patients.16
Acute Deep Venous Thrombosis
Venous thromboembolism (VTE) is a significant health care problem in this country, with an estimated 900,000 cases of deep venous thrombosis (DVT) and pulmonary embolism (PE), causing approximately 300,000 deaths yearly.17 For the past 150 years, understanding the pathogenesis of VTE has centered on Virchow’s triad of stasis, changes in the vessel wall (now recognized as injury), and thrombogenic changes in the blood. Stasis is probably permissive, and not a direct cause, whereas systemic infection and systemic inflammation may be more causal than previously thought.18,19
Venous Thrombosis Pathways
Hemostasis is typically initiated by damage to the vessel wall and disruption of the endothelium, although it may be initiated in the absence of vessel wall damage, particularly in venous thrombosis.20 Vessel wall damage simultaneously results in release of tissue factor (TF), a cell membrane protein, from injured cells and the circulation, with subsequent activation of the extrinsic pathway of the coagulation cascade. These two events are critical to the activation and acceleration of thrombosis. Tissues also vary with regard to their susceptibility to thrombosis, and the local organ mechanisms may be somewhat different. For example, hemostasis in cardiac muscle may be more dependent on the extrinsic pathway for thrombosis, whereas skeletal muscle may be more dependent on the intrinsic pathway for thrombosis.21
Platelet activation and the formation of an effective hemostatic “platelet plug” is a primary thrombotic event. Two platelet activation routes are thought to exist physiologically.22 Without direct vessel damage, platelet activation may occur via TF de-encryption and activation by protein disulfide isomerase, with factor VIIa generation and activation of platelets. Alternatively, subendothelial collagen may directly bind to glycoprotein (GP) VI and von Willebrand factor (vWF), leading to platelet capture and activation.
Platelet interactions and activation are mediated by vWF, whose receptor is GPIb, and via GPIIb/IIIa to fibrin.23Activation of platelets leads to the release of the prothrombotic contents of platelet granules, which contain receptors for coagulation factors Va and VIIIa. In addition, platelet activation also leads to the elaboration of arachidonic acid metabolites such as thromboxane A2, further promoting platelet aggregation (as well as vasoconstriction). Changes in platelet shape result in exposure of negatively charged procoagulant phospholipids normally located within the inner leaflet of the platelet membrane.24 Platelets also release microparticles (MPs), rich in TF and other procoagulants, which accelerate and concentrate the thrombus generation. Interestingly, circulating TF may be more important in venous thrombosis than in arterial thrombosis.20,25
The extrinsic pathway begins with activation of factor VII, by complexing TF with factor VII. The TF-VIIa complex then activates factors IX and X to IXa and Xa, respectively, in the presence of calcium. Feedback amplification occurs with factors VIIa, IXa, and Xa, all of which are capable of activating VII to VIIa, especially when bound to TF.26 Factor Xa is also capable of activating factor V to Va. Factors Xa, Va, and II (prothrombin) form on the platelet phospholipid surface in the presence of Ca2+ and initiate the prothrombinase complex, which catalyzes the formation of thrombin from prothrombin. Thrombin feedback amplifies the system by activating not only factor V to Va but also factors VIII (normally circulating bound to vWF) to VIIIa and XI to XIa. After activation, factor VIIIa dissociates from vWF and assembles with factors IXa and X on the platelet surface in the presence of Ca2+ to form a complex called the Xase complex, which catalyzes the activation of factor X to Xa.
Thrombin (factor II) is central to coagulation through its action of cleavage and release of fibrinopeptide A (FPA) from the α chain of fibrinogen and fibrinopeptide B (FPB) from the β chain of fibrinogen. This causes fibrin monomer polymerization and cross-linking, which stabilizes the thrombus and the initial platelet plug. Thrombin also activates factor XIII to XIIIa, which catalyzes the cross-linking of fibrin as well as that of other plasma proteins, such as fibronectin and α2-antitrypsin, resulting in their incorporation into the clot and increasing resistance to thrombolysis.27 In addition, factor XIIIa activates platelets as well as factors V and VIII, further amplifying thrombin production.
Coagulation can be activated through the intrinsic pathway with activation of factor XI to XIa, which subsequently converts factor IX to IXa and promotes formation of the Xase complex and ultimately thrombin. Another mechanism by which this occurs in vitro is through the contact activation system, whereby factor XII (Hageman factor) is activated to XIIa when complexed to prekallikrein and high-molecular-weight kininogen (HMWK) on a negatively charged surface; factor XIIa then activates factor XI to XIa. Both thrombin and factor XIa are also capable of activating factor XI.28 The physiologic importance of the intrinsic pathway is not completely clear and is probably not as physiologically important in the venous system as in the arterial system.
Natural Anticoagulants
Several interrelated processes localize thrombotic activity to sites of vascular injury. First, antithrombin (AT) is a central anticoagulant protein that binds to thrombin and interferes with coagulation by three major mechanisms: (1) inhibition of thrombin prevents removal of fibrinopeptides A and B from fibrinogen, limiting fibrin formation; (2) thrombin becomes unavailable for activation of factors V and VIII, thus slowing the coagulation cascade; and (3) thrombin-mediated platelet activation and aggregation are inhibited. In the presence of heparin, the accelerated inhibition of thrombin by antithrombin results in systemic anticoagulation. Antithrombin has been shown to directly inhibit factors VIIa, IXa, Xa, XIa, and XIIa. Thus, patients with a genetic deficiency of antithrombin are at much higher risk for development of VTE than the normal population is.
A second natural anticoagulant is activated protein C (APC), which is produced on the surface of intact endothelium when thrombin binds to its receptor, thrombomodulin, and endothelial protein C receptor (EPCR). The thrombin-thrombomodulin complex inhibits the actions of thrombin and also activates protein C to APC. APC, in the presence of its cofactor protein S, inactivates factors Va and VIIIa, therefore reducing Xase and prothrombinase activity.29
The third innate anticoagulant is tissue factor pathway inhibitor (TFPI). This protein binds the TF-VIIa complex, thus inhibiting the activation of factor X to Xa and formation of the prothrombinase complex. Interestingly, factor IX activation is not inhibited. Finally, heparin cofactor II is another inhibitor of thrombin whose action is in the extravascular compartment. The activity of heparin cofactor II is augmented by glycosaminoglycans, including both heparin and dermatan sulfate, but its deficiency is not associated with increased VTE risk.30
Physiologic Thrombolysis
Physiologic clot formation is balanced by controlled thrombolysis to prevent pathologic intravascular thrombosis. The central fibrinolytic enzyme is plasmin, a serine protease generated by the proteolytic cleavage of the proenzyme plasminogen. Its main substrates include fibrin, fibrinogen, and other coagulation factors. Plasmin also interferes with vWF-mediated platelet adhesion by proteolysis of GPIb.31
Activation of plasminogen occurs by several mechanisms. In the presence of thrombin, vascular endothelial cells produce and release tissue plasminogen activator (tPA) as well as α2-antiplasmin, a natural inhibitor of excess fibrin-bound plasmin. As clot is formed, plasminogen, tPA, and α2-antiplasmin become incorporated into it. In contrast to free circulating tPA, fibrin-bound tPA is an efficient activator of plasminogen.
A second endogenous activator of plasminogen is through the urinary plasminogen activator (uPA), also produced by endothelial cells but with less affinity for fibrin. Activation of uPA in vivo is not completely understood. However, it is hypothesized that plasmin in small amounts (produced through tPA) activates uPA, leading to further plasminogen activation and amplification of fibrinolysis.32
The third mechanism of plasminogen activation involves factors of the contact activation system; activated forms of factor XII, kallikrein, and factor XI can each independently convert plasminogen to plasmin. These activated factors may also catalyze the release of bradykinin from high-molecular-weight kininogen, which further augments tPA secretion. Finally, APC has been found to proteolytically inactivate plasminogen activator inhibitor type 1 (PAI-1), an inhibitor of plasmin activators that is released by endothelial cells in the presence of thrombin.33
The degradation of fibrin polymers by plasmin ultimately results in the creation of fragment E and two molecules of fragment D, which are released as a covalently linked dimer (D-dimer).34 Detection of D-dimer in the circulation is a marker for ongoing thrombus metabolism and has been shown to accurately predict ongoing risk of recurrent VTE.35
Interestingly, the resting state of the fibrinolytic system within the vein wall is lower in the area of the valvular cusps.36 In comparison with other anatomic locations, the deep veins of the lower limb have the lowest fibrinolytic activity in soleal sinuses as well as in the popliteal and femoral vein regions. This observation underlies a popular hypothesis as to why DVT most commonly originates in the lower limb. However, no in vivo real-time imaging studies have ever shown how and where DVT forms.
Plasminogen Inhibitors and Thrombosis
Activation of plasminogen provides localized proteolytic activity,37–39 and in plasma, PAI-1 is the primary inhibitor of plasminogen activators. It is secreted in an active form from liver and endothelial cells and is stabilized by binding to vitronectin (and inhibits thrombin in this form). PAI-1 is stored in the alpha-granules of quiescent platelets.40 PAI-1 levels are elevated by hyperlipidemia, and PAI-1 elevation appears to synergize with factor V Leiden genetic abnormalities.
Studies on the role of elevated PAI-1 in venous thrombosis have been contradictory,41,42 although it is plausible that elevated PAI-1 could suppress fibrinolysis and increase thrombosis potential. In humans, genetic polymorphisms correlate with increased risk of VTE. The highest levels of PAI-1 have been noted in those individuals carrying the 4G/4G polymorphism. Studies have found an eightfold higher risk for VTE in patients with the 4G allele in combination with other thrombophilic markers,43 and a 4.5-fold higher risk for pulmonary embolism in patients with 4G/4G polymorphism and protein S deficiency.44
Endothelium and Hemostasis
Most of the thrombosis-thrombolysis processes occur in juxtaposition to the endothelium, and hence the endothelium is one of the pivotal regulators of homeostasis (Fig. 12-2). Under normal conditions, endothelial cells maintain a vasodilatory and local fibrinolytic state in which coagulation, platelet adhesion, and activation are suppressed. A nonthrombogenic endothelial surface is maintained by a number of mechanisms, including (1) endothelial production of thrombomodulin and subsequent activation of protein C; (2) endothelial expression of heparan sulfate and dermatin sulfate, which accelerate antithrombin and heparin cofactor II activity; (3) constitutive expression of TFPI; and (4) local production of tPA and uPA. In addition, the production of NO and prostacyclin by the endothelium inhibits the adhesion and activation of leukocytes and produces vasodilation.45 TF production is also inhibited by NO.46
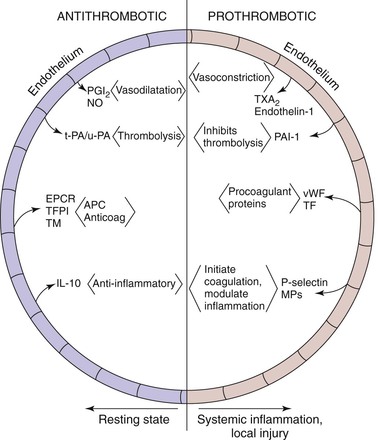
Figure 12-2 The careful balance between an antithrombotic and a prothrombotic milieu exists primarily at the endothelial level. Antithrombotic mediators include prostacyclin (PGI2) and nitric oxide (NO). Local thrombolysis is conferred by tissue (tPA) and urinary (uPA) plasminogen activators. Endothelial receptor for protein-C (EPCR), tissue factor pathway inhibitor (TFPI), and thrombomodulin (TM) inhibit thrombosis. Lastly, interleukin-10 (IL-10) is an anti-inflammatory cytokine. On the prothrombotic side, thromboxane (TXA2) and endothelin-1 promote vasoconstriction. Plasminogen activator inhibitor-1 (PAI-1) inhibits thrombolysis, and von Willebrand factor (vWF) and tissue factor (TF) are both procoagulant proteins. Lastly, P-selectin and microparticles (MPs) initiate coagulation and also modulate inflammation.
After endothelial injury a prothrombotic and proinflammatory state of vasoconstriction is supported by the endothelial surface. Release of platelet-activating factor (PAF) and endothelin-1 promotes vasoconstriction,47 whereas production of vWF, TF, PAI-1, and factor V augments thrombosis. Indeed, vWF is expressed to a greater extent on the endothelium of veins than on the endothelium of arteries, and tPA is less commonly expressed in venous endothelium.2 Systemic inflammatory insults such as conferred by tumor necrosis factor-α may cause endothelial activation and result in increased surface expression of cell adhesion molecules (CAMs) such as P-selectin, E-selectin, and intracellular CAM (ICAM), thereby promoting the adhesion and activation of leukocytes as well as platelets.7
Inflammation and Thrombosis
The relationship between thrombosis and inflammation was first suggested in the early 1970s.48 Inflammation increases TF, membrane phospholipids, fibrinogen, and the reactivity of platelets while decreasing thrombomodulin and inhibiting fibrinolysis (Fig. 12-3).49
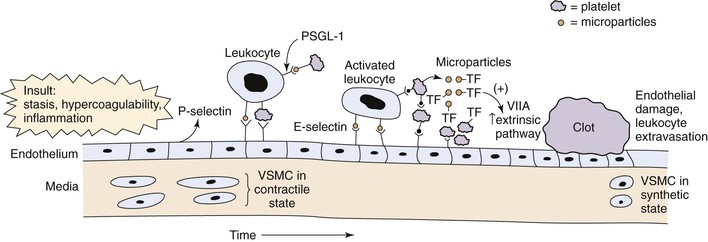
Figure 12-3 Acute venous thrombosis most commonly occurs in the lower extremity veins. Insults such as stasis, hypercoagulability, and inflammation result in damage to the endothelium and/or upregulation of P-selectin. Leukocytes bind via P-selectin glycoprotein ligand (PSGL-1) to P-selectin as well as platelets. This step leads to activated leukocytes, which then more firmly bind via E-selectin and release inflammatory mediators. Platelets also bind to leukocytes, endothelium, and other platelets and release microparticles that express tissue factor (TF). Tissue factor activates the extrinsic pathway by activation of factor VIIA (VIIA). A clot develops, and endothelial damage with further leukocyte adherence, extravasation, and inflammation occur. In the media, the normal state of the vascular smooth muscle cells (VSMCs) is contractile. Once activated and in the inflammatory state, the cells may become synthetic with production of profibrotic mediators and collagen.
Microparticles are involved in the initiation and amplification of thrombosis. They are small (<1 µm) phospholipid vesicles shed from platelets, leukocytes, and endothelial cells in a calcium-dependent fashion.50–52 Microparticles lack DNA and RNA, but subpopulations of microparticles rich in TF and phosphatidylserine have been identified.53,54 Fusion of microparticles with activated platelets results in decryption of TF and the initiation of thrombosis.55 Indeed, several circulating markers of inflammation once thought to be soluble are actually carried by microparticles.56–58 These vesicles have direct exogenous procoagulant activity, as shown by normalization of tail bleeding times in hemophilic mice.57 Moreover, microparticles shed from platelets express PAI-1, and these microparticles add to the growing thrombus via interactions between P-selectin and its receptor, P-selectin glycoprotein ligand-1 (PSGL-1). In this manner, platelet microparticles not only are prothrombotic but also inhibit fibrinolysis, facilitating thrombus growth.59
In veins, CAMs and microparticles interface in promoting thrombosis and inflammation. CAMs allow leukocyte transmigration, and selectins (P- and E-selectin) are integrally involved in venous thrombosis. P-selectin is upregulated in the vein wall as early as 6 hours after thrombus induction, and E-selectin has been found upregulated at later time points.60 Both venous stasis and hypoxia result in the upregulation of P-selectin, which localizes prothrombotic microparticles to the area of stasis and promotes the development of DVT.61–63 PSGL-1 is expressed on leukocytes and platelets as well as on their derived microparticles. Indeed, microparticles coexpressing TF and leukocyte markers have been shown to accumulate in growing thrombi in a P-selectin/PSGL-1–dependent fashion.64–66 Furthermore, P-selectin/PSGL-1 interaction stimulates the production of thrombogenic microparticles from leukocytes, along with platelets and endothelial cells.67
Although it was long thought to be a bystander in venous thrombosis unlike in the arterial system, the platelet is now thought to play a critical role.68 First, in stasis and nonstasis experimental murine venous thrombosis, genetic deletion of vWF was associated with significantly reduced size of venous thrombi that was not restored with recombinant factor VIII.69 Intravital microscopy also showed direct association of leukocytes and platelets in a growing acute thrombus. Consistently, platelets, via GPIb2, may promote venous thrombosis by colocalizing leukocytes and coagulation factors at the site of injury or stasis in the vein.70
Thrombus Resolution and Vein Wall Remodeling
Regardless of the location and extent of acute DVT, the resolution process is complex71–73 (Table 12-2 and Fig. 12-4). In humans, the natural fibrinolytic mechanisms break down the thrombus over time and at variable rates.74,75 Resolution of experimental venous thrombus resembles wound healing, involving profibrotic growth factors, collagen deposition, and activation of matrix metalloproteinase (MMP). The fact that leukocytes invade the thrombus in a specific sequence suggests their importance in normal thrombus resolution.76,77 Real-time in vivo imaging has also shown that leukocyte concentration and MMP activity colocalize to areas of recanalization.48,78,79
Table 12-2
Experimental Venous Thrombosis Resolution Characteristics
| Acute (<8 Days) | Chronic (>8 Days) | |
| Effector leukocyte | Neutrophil | Monocyte |
| Chemokines/cytokine | IL-8, IL-1β, IL-6 | Monocyte chemotactic protein-1 Secondary lymphoid chemokine |
| Growth factors | — | Vascular endothelial growth factor Basic fibroblast growth factor, transforming growth factor–β |
| Neovascularization of clot | Minimal | Yes von Willebrand factor–positive channels, hypoxia-inducible factor-1α |
| Matrix remodeling | MMP-9, elastase? | Urinary plasminogen activator–plasmin MMP-2, plasminogen activator inhibitor-1–vironectin |
| Vein wall, collagen type | Collagen III | Collagen I |
IL, Interleukin; MMP, matrix metalloproteinase.
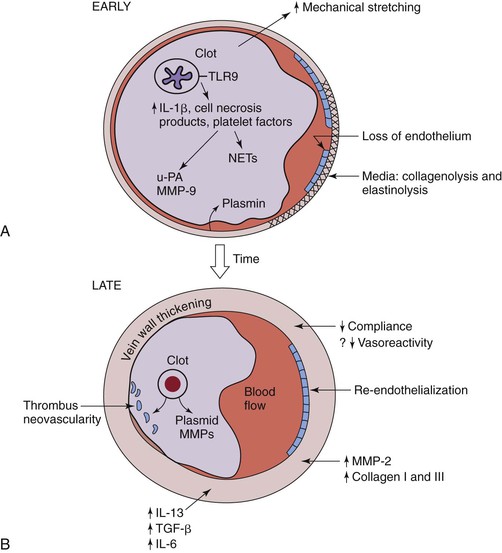
Figure 12-4 Hypothesized thrombosis resolution, based on small rodent models, is shown. A, Early thrombus resolution involves a large clot releasing interlukin-1β (IL-1β), cell necrosis products, and platelet factors that drive neutrophil influx, with release of plasminogen activators, such as urinary platelet activator (uPA). Concurrently, matrix metalloproteinase-9 (MMP-9) is released, and plasmin is upregulated. This allows for early thrombolysis. At the same time, dying polymorphonuclear neutrophils (PMNs) may contribute to thrombosis with release of neutrophil extracellular traps (NETs). Concurrently, loss of endothelium exposes the subendothelial matrix proteins that may further potentiate thrombosis. In the media, collagenolysis and elastinolysis occur in addition to direct mechanical stretch. B, Later (usually after 8 days), vein wall medial thickening occurs with decreased compliance and, possibly, decreased vasoreactivity. Reendothelization commences but is incomplete until a much later time point (>14 days). Thrombus neovascularity and cellularity of the thrombus are associated with resolution, which is a monocyte/macrophage-driven process. Within the vein wall, matrix turnover occurs with increased MMP-2 expression, as well as collagen I and collagen III production. IL-13 and transforming growth factor-β (TGF-β) are two profibrotic growth factors that may be involved with late vein wall remodeling.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


