Chapter 3 Vascular Smooth Muscle
With the evolution of an enclosed circulatory system to transport oxygenated blood, hormones, immune cells, metabolites, and waste products to and from cells in distal sites within the vertebrate body, blood vessels evolved adaptations necessary for repeated cycles of contraction and extension resulting from cardiac-driven pulsatile blood flow. These adaptations for blood vessel distensibility allow elastic conductance arteries in the macrocirculation, under the influence of the pulsatile cardiac cycle, to provide blood flow to end organs by altering the luminal diameter of the vessel. They also allow resistance arteries in the microcirculation, which experience steady flow, to regulate vasomotion at the organ level to maintain blood pressure homeostasis.1 The cells that primarily establish and orchestrate these contraction and distensible properties are vascular smooth muscle cells (VSMCs), the majority cell type within the normal vessel wall. VSMCs maintain contractile tone by a highly organized architecture of contractile/cytoskeletal proteins and associated regulatory components within the cell cytoplasm and establish distensibility by synthesis, secretion, and organization of extracellular matrix (ECM) components with elastic recoil and resilience properties.1 VSMCs within the vascular continuum have the ability to adapt expression of proteins involved in contraction and ECM synthesis according to extrinsic and intrinsic cues during different developmental stages and in disease or response to injury. This ability is due to a phenomenon known as VSMC phenotypic modulation and is a major feature that distinguishes VSMCs from terminally differentiated cells.2
Vascular smooth muscle cell phenotypic modulation is the ability to switch phenotypic characteristics from a migratory synthetic phenotype in embryonic tissue patterning to a quiescent, contractile phenotype in maintenance of vascular tone in mature vessels. Importantly, during vascular remodeling in response to injury, VSMCs can switch back to a synthetic phenotype characterized by increased VSMC proliferation and ECM synthesis. Although the ability to switch phenotypes may have evolved as an adaptive survival mechanism for VSMCs to adjust physiological responses due to changing hemodynamic demands or to repair damage after vascular injury, phenotypic modulation has important implications both during development and during vascular disease.2
This chapter will highlight how these diverse functions of VSMCs arise from both innate genetic programs and a range of diverse environmental cues that include soluble signaling factors, insoluble ECM components, physical mechanical forces, and interactions with other cell types.3 Discussion will center on the complex webs of signaling networks generated by these diverse external factors, and how these networks are regulated and integrated at multiple transcriptional and posttranslational levels to mediate the diverse functions of VSMCs in normal physiology and disease/injury pathology.
Origins of Vascular Smooth Muscle Cells During Embryonic Development
Initially in embryonic vasculature development, endothelial precursor cells form a common progenitor vessel which then gives rise to the first artery (dorsal aorta) and vein (cardinal vein) by selective sprouting and subsequent arterial-venous cell segregation4 (see also Chapter 1). The distinct molecular identities of arteries and veins are regulated by complex interactions of several signaling pathways, including sonic hedgehog (Shh), a member of the hedgehog (Hh) family of secreted morphogens; secreted growth factors in the vascular endothelial growth factor family (VEGFs)5; Notch receptors (Notch 1-4) and Notch ligands (Jagged1,2); and transmembrane proteins that can transduce cell-cell interactions into signals determining cell fates.6 Interactions of these signals induce differential expression of VEGF receptors, Ephrin ligands, and tyrosine kinase Eph receptors on the segregating arterial/venous cells, with ephrin B2 and EphB4 as markers expressed in arteries and veins, respectively.4,5 In response to VEGF signaling, endothelial cells (ECs) within these primordial vascular networks recruit mural cells, including nascent VSMCs.7
Nascent VSMCs derive from multiple and nonoverlapping embryonic origins that are reflected in different anatomical locations within the adult. Ectodermal cardiac neural crest cells give rise to the large elastic arteries (e.g., ascending and arch portions of the aorta), ductus arteriosus, and carotid arteries; proepicardium mesothelial cells produce the coronary arteries; mesodermal cells are origins for the abdominal aorta and small muscular arteries; the mesothelium forms the mesenteric vasculature; secondary heart field cells form the base of the aorta and pulmonary trunk; somite-derived cells produce the descending thoracic aorta; and satellite-like mesoangioblasts give rise to the medial layers of arteries.8 The heterogeneous mosaic of VSMCs in the vessel wall may be due in part to these diverse embryological origins of VSMCs and could be reflected in the presence of phenotypically distinct subpopulations within the media that account for VSMC plasticity.9 There is some evidence that VSMCs derived from different lineages exhibit morphologically and functionally distinct properties and respond differently to soluble factors in vitro and to morphogenetic cues in vivo,8 suggesting that the major determinants of VSMC responses to signals in vascular development are principally lineage-dependent rather than environment-dependent.8
Vascular Smooth Muscle Cell Phenotypic Modulation
Characterization of Vascular Smooth Muscle Cell Phenotypes
Given the multiple origins and distinct subpopulations of VSMCs, a compelling central question for understanding VSMC biology is how cells from these diverse embryonic origins, initially expressing lineage-specific pathways, differentiate to express the same marker genes specifically characteristic of VSMCs.8,10 Another question is how these same VSMCs, responding to both extrinsic and intrinsic cues, can alter expression of these genes (and thus molecular pathways), leading to diverse phenotypes with distinct and diverse functions. VSMC phenotypes can be loosely divided into three types: contractile/differentiated, synthetic/dedifferentiated, and inflammatory.
Contractile, differentiated vascular smooth muscle cells
Contractile or differentiated VSMCs are characterized by a repertoire of contractile proteins, contractile-regulating proteins, contractile agonist receptors, and signaling proteins responsible for contraction and maintenance of vascular tone.3,11,12 Of the VSMC “marker” proteins expressed in the contractile phenotype repertoire (Fig. 3-1), the most discriminating markers are smooth muscle myosin heavy chain (SMMHC) in conjunction with alpha-smooth muscle actin (αSMA), smoothelin, SM-22α, h1-calponin, and h-caldesmon.2 In addition to expressing these proteins associated with contractile function, contractile VSMCs exhibit differential levels of ECM components (increased collagen types 1 and IV) and matrix-modifying enzymes (decreased matrix metalloproteinases [MMPs] and increased tissue inhibitors of matrix metalloproteinases [TIMPs]). Contractile VSMCs are further characterized by an elongated spindle-shaped morphology in culture, a low proliferative rate, and expression of α1β1, α7β1 integrins and the dystrophin-glycoprotein complex (DGPC).3,13
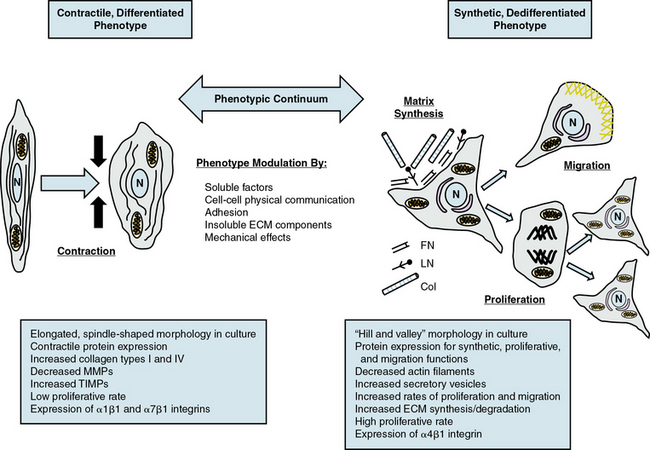
Synthetic, dedifferentiated vascular smooth muscle cells
Synthetic or dedifferentiated VSMCs have decreased expression of SMC-related genes for contractile proteins (e.g., SMMHC), with concomitant increased osteopontin, l-caldesmon, nonmuscle myosin heavy chain B, vimentin, tropomyosin 4, and cellular-retinal binding-protein-1 (CRBP-1) (see Fig. 3-1). “Positive” marker genes, such as nonmuscle myosin heavy chain (NM-B MHC) or SMMHC embryonic (SMemb) expressed specifically in embryonic or phenotypically modified VSMCs, are characteristic of dedifferentiated VSMCs in association with vascular injury.2 Other characteristics of synthetic VSMCs include decreased number of actin filaments, an increase in secretory vesicles, increased rates of proliferation and migration, extensive ECM synthesis/degradation capabilities, increased cell size and “hill-and-valley” morphology in culture, high proliferative rate, and increased expression of α4β1 integrin.
Inflammatory vascular smooth muscle cells
In addition to the phenotypic continuum between contractile and synthetic phenotypes, VSMCs can also express markers of an inflammatory phenotype in response to EC-induced recruitment of monocytes and macrophages during the progression of atherosclerosis.14 Various stimuli, including secretion of cytokines by these inflammatory cells, changes in ECM composition, oxidized low density lipoprotein (oxLDL), and VSMC interactions with monocytes/macrophages, induce expression of inflammatory cytokines, vascular cell adhesion molecule (VCAM-1) and transcription factors (NFκB) in VSMCs, leading to recruitment of inflammatory cells into the vessel wall.
Upstream Mediators of Phenotypic Modulation
Growth-inducing factors
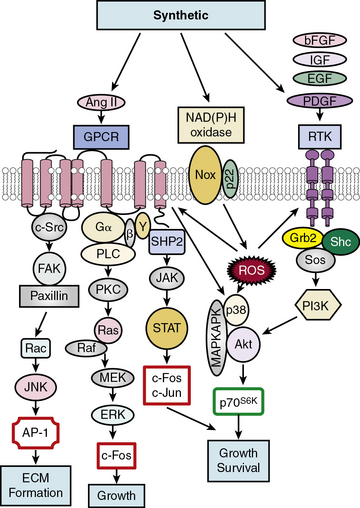
Soluble factors that include growth factors, hormones, and reactive oxygen species (ROS) serve as upstream mediators of the phenotypic switch from contractile to synthetic VSMCs, which results in large part from coordinate activation/repression of VSMC marker genes important in the contractile response2,3,15,16 (Fig. 3-2). Some of the most important growth-inducing factors include platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin-like growth factor (IGF), and basic fibroblast growth factor (bFGF). Growth factors bind to surface membrane receptor tyrosine kinases (RTKs), triggering sequential downstream signaling pathways mediated through complex formation of activated RTKs with adaptor and signaling proteins Grb2/Shc/Sos, and activation of intracellular kinases, including phosphatidylinositol 3-kinase (PI3K), mitogen-activated protein kinases (MAPKs: extracellular signal regulated kinase, ERK1/2, p38MAPK, and c-jun NH2-terminal kinase, JNK), Akt, MAPKAPK2, and p70S6 kinase (p70S6K). These signals not only transcriptionally mediate the switch to the synthetic phenotype, but also serve to promote growth and survival. In addition, ROS such as hydrogen peroxide (H2O2) produced by activation of NADPH oxidases, multimeric enzymes containing p22phox and other subunits depending upon the specific isoform, can act as second messengers for canonical G protein–coupled receptor (GPCR) and RTK pathways.17
Differentiation-inducing factors
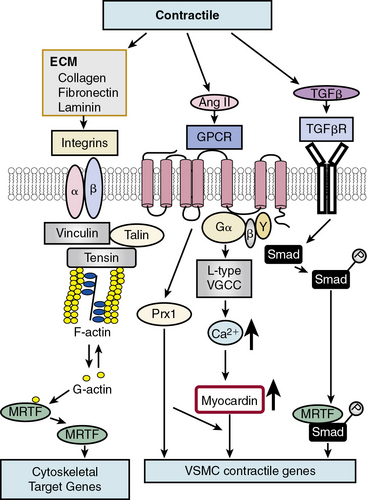
In contrast to growth factor–stimulated proliferation, the cytokine transforming growth factor β (TGF-β) and members of the bone morphogenetic protein (BMP) subgroup of this family promote the differentiated, contractile phenotype in VSMCs by inducing expression of the VSMC contractile genes αSMA and calponin (Fig. 3-3). Transforming growth factor β binds to a tetrameric complex consisting of two type I and two type II receptors, resulting in phosphorylation of Smads, transcription factors named for Caenorhabditis elegans Sma and Drosophila Mad (mothers against decapentaplegic).18 Within the TGF-β signaling pathway itself, different Smads control expression of different markers. For example, Smad3 transactivates the SM22α promoter, while Smad2 activates the αSMA gene. Other soluble factors that inhibit proliferation and increase differentiation include heparin and retinoic acid.9 Most smooth muscle differentiation markers share additional common transcriptional pathways, discussed in more detail later. For example, both TGF-β-induced phosphorylated Smads and ECM-induced activation of integrins, mediated through focal adhesion components vinculin, talin, and tensin, in concert with changes in cytoskeletal F/G actin dynamics, result in myocardin-related transcription factor (MRTF) induction of cytoskeletal/contractile genes (see Fig. 3-3).
Dual factors
One factor with a potential dual role, depending upon initial phenotype/developmental stage, is the octapeptide hormone angiotensin II (Ang II), the effector molecule of the renin–angiotensin II system.19 Angiotensin II can induce either contractile or synthetic phenotypes, with differential responses depending upon cell context and locations within the artery (see Figs. 3-2 and 3-3). Angiotensin II, binding to its GPCR AT1R, activates VSMC marker gene expression indicative of the contractile phenotype through L-type voltage-gated Ca2 + channel–induced elevations in intracellular Ca2 + concentrations, and subsequent increased myocardin transcription coactivator expression dependent upon Prx1, a homeodomain protein that promotes serum response factor (SRF) binding to conserved elements in VSMC marker gene promoters.20 In addition, Ang II binding to AT1R can induce signatures of the synthetic phenotype by activation of multiple kinase and enzyme pathways that are interconnected in signaling networks (see Fig. 3-2). These include the MAPKs; RTKs, including ROS-sensitive transactivation of epidermal growth factor receptor (EGFR); nonreceptor tyrosine kinases (c-Src/focal adhesion kinase [FAK]/paxillin/Rac/JNK/AP-1) and tyrosine phosphatases; SHP2/Janus kinase and signal transducers and activators of transcription (JAK/STAT); and GPCR classic signaling cascades (phospholipase C [PLC]/protein kinase C [PKC]/Ras/Raf/mitogen extracellular signal regulated kinase [MEK]/ERK) leading to stimulation of early growth-response genes (c-fos, c-jun), survival pathways (e.g., Akt), and ECM formation (JNK/AP-1).
Notch communication
In addition to its critical function in development, Notch signaling is also important in defining VSMC differentiation.21,22 Downstream Notch effector gene activation results in activation of “master regulators” of VSMC differentiation (myocardin, MRTFs, or SRF) or direct induction of contractile proteins SMMHC and αSMA, as well as the VSMC specific differentiation marker SM22α (also known as transgelin).6 Data regarding Notch signaling on VSMC differentiation, however, are conflicting, with some studies supporting a repressive effect, while others indicate a promoting effect on expression of VSMC marker genes SMMHC and αSMA.22 These discrepancies may be due to the antagonistic roles of Notch and the Notch effector Hairy-related transcription factor 1 (HRT1) on markers of VSMC differentiation, specifically αSMA and SMMHC.23 Hairy-related transcription factor 1 inhibits Notch/RBP-Jκ binding to the αSMA promoter in a histone deacetylase-independent manner. The context-dependent roles of Notch and HRT1 on markers of VSMC differentiation may serve to fine-tune VSMC phenotypic modulation during vascular development, injury, and disease.
There is considerable cross-talk between Notch and other signaling pathways. Notch and TGF-β cooperatively induce a functional contractile, differentiated phenotype through parallel signaling axes,24 while HRT factors block VSMC differentiation in both pathways. Other examples of cross-talk among key signaling pathways for morphogenesis (Hh, Notch) and mitogenesis (VEGF-A, PDGF) include a Shh/VEGF-A/Notch signaling axis in VSMCs in the neointima to increase growth and survival,25 and Notch-induced up-regulation of PDGFR-β to mediate growth and migration.26
Homotypic VSMC-VSMC Notch-mediated signaling pathways are also apparent in adult vascular pathologies and response to injury.22 After injury, Notch receptors are increased, along with elevated levels of HRT. Negative feedback between HRT and Notch may account for the adaptive response to injury in which initial Notch/HRT-induced suppression of the contractile phenotype is followed by arterial remodeling. As Notch/HRT signaling decreases, the contractile phenotype is reestablished.24
Transcriptional Regulation of Vascular Smooth Muscle Cell Diversity
The complex web of signaling pathways induced by these external signals—whether they are soluble, insoluble, structural, or mechanical—converge on a network of transcription factors (TFs) that coordinately regulate gene expression and act as “master switches” for growth and differentiation27 (Fig. 3-4). Transcription of VSMC-specific differentiation or proliferative genes is regulated by cooperative interaction of TFs and their coregulators, including SRF,28 myocardin and myocardin-related TFs (MRTF-A and -B),29 Ets domain transcription factors known as ternary complex factors (TCFs),30 zinc finger factors GATA630 and PRISM/PRDM6,31 and Krüppel-like factors (KLFs).32,33
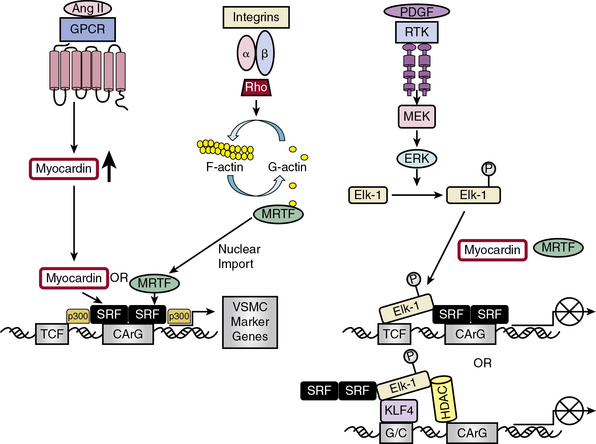
Serum response factor/myocardin axis
Serum response factor, a widely expressed member of the MADS (MCM1, agamous, deficien, SRF) box of TFs, is a nodal point linking signaling pathways to differential gene expression related either to growth or differentiation, depending upon which transcriptional partner is bound to SRF.28 Serum response factor self-dimerizes and binds with high affinity and specificity to a consensus deoxyribonucleic acid (DNA) sequence CArG box found in the promoters of cyto-contractile genes.34 More than half of the VSMC “marker” genes that define VSMC molecular signature contain CArG boxes.34 Included in these genes are three categories modulating actin filament dynamics: (1) structural (e.g., αSMA-actin, SM22α, caldesmon, SMMHC); (2) effectors of actin turnover (e.g., cofilin, gelsolin); and (3) regulators of actin dynamics (four-and-a-half LIM domains proteins [FHL1 and 2], MMP9, and myosin light chain kinase).35
Serum response factor itself is a weak activator of CArG-dependent genes.30 Potent SRF-dependent transcriptional activation is therefore dependent upon regulation at several levels: by interaction with different signal-regulated or tissue-specific regulatory SRF transcription cofactors/corepressors; by posttranslational phosphorylation, acetylation, and sumoylation, modifications that affect these interactions; and by epigenetic alterations in chromatin structure in which myocardin serves as a scaffold for recruitment of chromatin-remodeling enzymes36 that enable SRF and its cofactors to gain access to SRF target genes.8 Myocardin association with histone acetyltransferases (HATs), including p300, enhances transcription of VSMC-restricted genes, whereas association with class II histone deacetylases (HDACs) suppresses myocardin-induced transcription of VSMC marker genes36 (see Fig. 3-4).
Serum response factor interacts with cofactors in two principal families: the TCF family of Ets-domain proteins (Elk, SAP-1, and Net)37 activated by the MAPK pathway, leading to SRF binding to immediate early growth factor-inducible genes such as c-fos28; and the myocardin/MRTF-A/MRTF-B family35 to promote activation of VSMC-specific marker genes, most of which code for filamentous proteins that function in contractile activities or proteins that function in cell-matrix adhesions.10 These alternative pathways provide the “plasticity” associated with VSMC phenotypic modulation ranging from contractile functions to maintain vascular tone to synthetic or proliferative functions in response to vascular injury.29
Discovery of the cell-restricted SRF transcriptional coactivator myocardin, expressed specifically in cardiac and VSMCs, resolved the paradoxical observations that SRF can regulate mutually exclusive gene expression programs for growth or differentiation.30,34 In VSMCs, myocardin is a master regulator of SMC marker gene expression and sufficient for the smooth muscle–like contractile phenotype. Myocardin competes with Elk-1 for direct binding to SRF in VSMCs; thus, myocardin and Elk-1 can act as binary transcriptional switches that may regulate contractile vs. synthetic VSMC phenotypes30 (see Fig. 3-4). In addition, myocardin transduction leads to lower levels of the cell cycle–associated gene cyclin D1, resulting in repression of growth. Therefore, myocardin is a nodal point for two features indicative of SMC differentiation: expression of the contractile apparatus and suppression of growth.30
While myocardin functions exclusively as a transcriptional coactivator,38 additional proteins function to regulate transcriptional activity of myocardin. Hairy-related transcription factor 2 and GATA factors repress or enhance myocardin-induced transcriptional activity, depending upon cell context.30 In addition, activation of Notch receptors by Jagged1 endogenous ligand induces translocation of Notch intracellular domain (ICD) to the nucleus where it inhibits myocardin-induced SMC gene expression.29 Angiotensin II stimulation, as well as activation of L-type voltage-gated Ca2 + channels, activates SMC marker genes by inducing myocardin expression and, in the case of Ang II, increasing SRF binding to CArG elements in the promoter regions of VSMC marker genes such as αSMA.20
Serum response factor transcriptional activity is also controlled by Rho-induced actin dynamics that facilitate movement of MRTFs into or out of the nucleus29 (see Fig. 3-4). In most cell types, MRTFs form a stable complex with monomeric G-actin and remain sequestered in the cytoplasm. Myocardin-related transcription factors in VSMCs, however, are localized in the nucleus where binding to SRF in the basal state promotes contractile gene expression, and the differentiated phenotype. In response to growth factors or vascular injury, extracellular signals transduced through the Rho-actin pathway result in nuclear export of MRTF, down-regulation of SRF/MRTF-induced VSMC contractile gene expression, and promotion of mitogen-induced ERK1/2 phosphorylation of TCFs, resulting in TCF displacement of MRTFs and SRF/TCF-mediated activation of growth-responsive genes.29 These differential pathways provide a switch in which SRF target genes are differentially regulated through growth factor–induced signaling for growth (active TCF, MRTF blocked) or Rho-actin signaling for differentiation (inactive TCF, MRTF active)30 (see Fig. 3-4).
Zinc finger proteins
GATA6, a zinc finger transcription factor expressed in VSMCs, induces growth arrest by increasing expression of the general cyclin-dependent kinase inhibitor (CDKI) p21CIP1 and inhibiting S-phase entry.30 PRISM is a smooth muscle–restricted member of zinc finger proteins belonging to the PRDM (PR domain in smooth muscle) family and acts as a transcriptional repressor by interacting with class I histone deacetylases and G9a histone methyltransferases. PRISM induces the proliferative phenotype while repressing differentiation regulators myocardin and GATA6.31
One of the most intensely studied zinc finger transcriptional regulators in VSMCs is the KLF subfamily that binds to the TGF-β control element (TCE) in the regulatory sequences of target genes (reviewed in32,33,39). Vascular smooth muscle cells express four KLFs (KLF4, KLF5, KLF13, and KLF15), each with individual biological functions implicated in regulating a range of processes in both growth and differentiation.32 Individual KLFs may have opposing functions, depending upon temporal and developmental expression patterns and interactions with other factors. For example, KLF4 inhibits, whereas KLF5 and KLF13 induce, VSMC marker gene expression. Mechanisms that may account for these opposing functions of KLF factors include posttranslational modifications, interaction with specific cofactors, differential expression by growth factors, cytokines and differentiation state, or regulation by another KLF.32
KLF4 functions as both a VSMC growth repressor and a repressor for VSMC differentiation, although data on the effect of KLF4 on VSMC differentiation are conflicting33 (see Fig. 3-4). As a growth repressor, KLF4 inhibits PDGF-BB-induced mitogenic signaling and induces expression of the negative cell cycle regulator p53 and its target gene p21CIP1. As a differentiation repressor, KLF4 prevents SRF from binding to the TCE in promoters of VSMC marker genes, suppresses expression of myocardin, inhibits myocardin-induced activation of SMC marker genes, reduces SRF binding to CArG elements in SMC contractile gene promoters,33 and induces histone hypoacetylation at SMC CArG regions associated with gene silencing.40 On the other hand, there is evidence that KLF4 promotes VSMC differentiation by directly activating VSMC marker gene transcription of SM22α and αSMA.33 KLF4 thus functions as a bifunctional TF or “molecular switch” that can both activate and repress VSMC marker genes, depending upon regulation of KLF4.33
Even though the closely homologous KLF4 and KLF5 TFs share similar developmental and tissue pattern expression, they exert different, often opposing, effects on gene regulation and proliferation/differentiation.33 Whereas KLF4 is associated with growth arrest, KLF5 exerts pro-proliferative effects, particularly in vascular remodeling in response to injury. KLF5 expression, abundant in fetal VSMCs but down-regulated in the adult (reviewed in39), is induced after vascular injury by activation of immediate early response genes by Ang II and ROS.41 KLF5 in turn mediates re-expression of SMemb/NMHC-B, a marker for the dedifferentiated phenotype, and activates other critical injury response genes involved in remodeling, such as PDGF-A/B, Egr-1, plasminogen activator inhibitor 1 (PAI-1), inducible nitric oxide synthase (iNOS), and VEGFR, implicating KLF5 as a key regulator for VSMC response to injury.39 In additional injury responses, KLF5 increases cyclin D1 expression and inhibits the cyclin kinase inhibitor p21, thus leading to vascular remodeling by increased cell proliferation.42 Similar to KLF4 regulation, KLF5 expression and activity are regulated at multiple levels, including upstream Ras/MAPK, PKC, and TGF-β signaling pathways; downstream interactions with TFs, including retinoic acid receptor α (RARα), NF-κB, and peroxisome proliferator–activated receptor gamma (PPARγ); as well as posttranslational modifications that can positively or negatively regulate KLF activity.39 In addition, KLF5 activity is regulated in the nucleus by chromatin remodeling factors such as SET, a histone chaperone that inhibits the DNA binding activity of KLF5,43 p300 (a coactivator/acetylase that coactivates KLF5 transcription), and HDAC1, which inhibits KLF5 binding to DNA.32
Two additional KLFs have been identified in VSMCs: KLF13 and KLF15.32 After vascular injury, KLF13 is induced and activates the promoter for the VSMC differentiation marker SM22α, while KLF15 expression is down-regulated, implicating KLF15 as a negative regulator of VSMC proliferation and a counterbalance to the growth-promoting effects of KLF5 in vascular injury response.
Posttranscriptional Regulation of Vascular Smooth Muscle Cell Diversity: Noncoding microRNAs
Upstream signaling and downstream transcriptional pathways in VSMCs are intertwined with a multitude of micro ribonucleic acid (miRNAs) that act as “rheostats” and “switches” in regulating protein activity in development, function, and disease.44 miRNAs are small, noncoding RNAs (20-25 nucleotides in length) that associate with a miRNA-induced silencing complex (miRISC) of regulatory proteins, including Argonaute family proteins, Argonaute interacting proteins of the GW182 family, eukaryotic initiation factors (eIFs), polyA-binding complexes, decapping enzymes/ activators, and deadenylases, to induce posttranscriptional silencing of their target genes.45 These multiple components are assembled and interact in a multistep process with components of the translational machinery to inhibit translation initiation, mark mRNAs for degradation through deadenylation, and sequester targets into cytoplasmic P bodies.44 Multiple mechanistic models for miRNA-induced gene silencing have been proposed that provide insights into the molecular mechanisms of translational inhibition, deadenylation, and mRNA decay, but questions remain concerning the kinetics and ordering of these translational events and whether they are coupled or independent.45 A recent unifying model for miRNA-regulated gene repression is an attempt to reconcile the often conflicting existing data. It proposes that recruitment of Argonaute and associated GW182 proteins to miRNA induces binding to the mRNA 5′m7 Gcap, thus blocking translation initiation, potentially by mRNA deadenylation. Subsequent to miRNA-mediated deadenylation, mRNA is degraded through recruitment of decapping proteins.46 In this model, inhibition of translation initiation is linked to subsequent rapid mRNA decay in a coupled process. Because miRNAs—which in general are negative regulators of gene expression—may be almost as important as transcription factors in controlling gene expression in the pathogenesis of human diseases,47 insights into the functions of this class of noncoding RNAs are important in evaluating their potential use as therapeutic targets.45
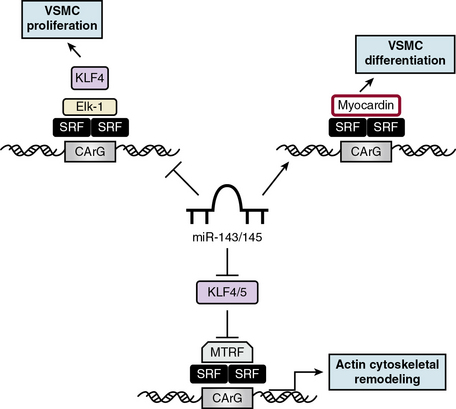
Cardiovascular-specific, highly conserved miRNAs miR-143 and miR-145, the most abundant miRNA in the vascular wall,48 are key players in programming VSMC fate from multipotent progenitors in embryonic development and in reprogramming VSMCs during phenotypic modulation in the adult44,49 (Fig. 3-5). miR-143 and miR-145 have distinct sequences but are clustered together and transcribed as a bicistronic unit. Upstream in the genomic sequence of miR-143/145 is a conserved SRF-binding CArG box site, indicating control by SRF and myocardin.49,50 These miRNAs cooperatively feed back to modulate the actions of SRF by targeting a network of transcription factors/coactivators/corepressors. This network includes miR-145-induced repression of KLF4, a positive regulator of proliferation and myocardin repressor; miR-143-induced repression of Elk-1, a myocardin competitor and positive regulator of proliferation; and, contrary to the usual inhibitory role of miRNA, miR-145-induced stimulation of myocardin, a positive regulator of differentiation. Thus, miR-145 is necessary and sufficient for VSMC differentiation, and the miR-143/miR-145 cluster acts as an integrated signaling node to promote differentiation while concurrently repressing proliferation.49 Although mice with genetic deletions for miR-143/145 show no obvious abnormalities in early development, VSMCs in the adult exhibit both structural and phenotypic differences in injury- or stress-induced vascular remodeling. Ultrastructural analysis of arteries from miR-143/145 knockout mice shows reduced numbers of medial VSMCs with a contractile appearance, and an increase in synthetic VSMCs.51 These results suggest that miR-143 and miR-145 modulate cytoskeletal structure, actin dynamics, and modulation to a dedifferentiated phenotype50 (see Fig. 3-5). Importantly, miR-143/145 knockout mice with increased synthetic VSMCs develop spontaneous neointimal lesions in the femoral artery in the absence of hyperlipidemia and inflammation, supporting a key role for phenotypically altered VSMCs in the pathogenesis of lesion formation.51
While miR-143 and miR-145 play keys roles in the contractile phenotype of VSMCs and the response to injury,52 miR-221 and miR-222 are modulators of VSMC proliferation, although largely by affecting growth-related signaling pathways rather than by controlling VSMC phenotype. miR-221 and miR-222, encoded by a gene cluster on the X chromosome, are up-regulated in VSMCs in neointimal lesions and in proliferating cultured VSMCs stimulated by PDGF-BB.53 Studies show that two CDKIs, p27KIP1 and p57KIP2, have miR-221 and miR-222 binding sites and are gene targets for miR-221 and miR-222 in the rat carotid artery in vivo.53 Thus, miR-221 and miR-222 are pro-proliferative because they repress two CDKIs, p27KIP1 and p57KIP2. Furthermore, PDGF, via miR-221 induction, inhibits VSMC differentiation via c-kit-induced inhibition of myocardin.54
Posttranslational Regulation of Vascular Smooth Muscle Cell Diversity: Epigenetics
The “epigenetic landscape” controls gene expression by chemical modifications that mark regions of chromosomes either by methylation of promoter CpG sequences in the DNA itself, or by covalent modification of histone proteins that package DNA by posttranslational addition of methyl, acetyl, phosphoryl, ubiquityl, or sumoyl groups, leading to expression/repression of transcription (reviewed in55). In VSMCs, multiple levels of epigenetic controls exist for gene expression leading to differentiation or dedifferentiation programs in healthy cells and for dysregulated gene expression in vascular disease. These epigenetic changes in VSMCs involve both DNA and histone methylation as well as histone acetylation/deacetylation. Methylation of histones, catalyzed by histone methyltransferases (HMTs), results in a tight, stable epigenetic mark between methylated histones and chromatin that can be passed to daughter cells, thus providing “epigenetic memory” that defines cell lineage and identity by controlling SRF access to VSMC-specific marker genes.55 Acetylation is controlled by HATs, which promote gene transcription by destabilizing chromatin structure to an “open,” transcriptionally active conformation, and HDACs, which promote chromatin condensation to a “closed,” transcriptionally silent conformation with restricted access to DNA. Histone acetylation/deacetylation thus serves to regulate transcription in a rapid and “on/off” manner in response to dynamic environmental changes and links the cell’s genome with new extrinsic signals.55
In VSMCs, SRF binding to CArG boxes in the promoters of SMC marker genes to promote the VSMC differentiated phenotype depends upon alterations of chromatin structure, including histone methylation and acetylation. In a model for epigenetic regulation of VSMC phenotype,56 SRF binding to CArG boxes in VSMC marker gene promoters is blocked by conditions such as PDGF-BB exposure or vascular injury. Such conditions promote KLF4-induced myocardin suppression as well as KLF4-induced recruitment of HDACs, resulting in “closed” deacetylated chromatin and transcriptional repression of VSMC marker genes. Histone methylation, in contrast, is not affected by PDGF-BB and may serve as a permanent “memory” for VSMC identity during repression of SRF-dependent transcription and can, once repressive signals are terminated, reactivate the differentiation program by recruiting myocardin/SRF complexes or HATs to VSMC marker genes for reexpression. In the absence of KLF4 activation, SRF/myocardin can bind to HAT-induced acetylated “open” chromatin at CArG boxes for transcriptional activation of VSMC marker genes, thus promoting VSMC differentiation. In addition, myocardin induces acetylation of histones in the vicinity of SRF-binding promoters in VSMC marker genes by association with p300, a ubiquitous transcriptional coactivator with its own intrinsic HAT activity, leading to synergistic activation of VSMC marker gene expression. This pro-myogenic program is antagonized and repressed by myocardin binding to class II HDACs, which strongly inhibits expression of marker genes αSMA, SM22α, SMMLCK and SMMHC. These opposing actions of HATs and HDACs on SRF/myocardin function to activate or repress, respectively, VSMC differentiation and serve to regulate transcription in a rapid and reversible manner in response to dynamic changes in the environment.55
Often, transcription mediators play roles in both classic signal transduction pathways and epigenetic programming.57 Smad proteins, for example, transmit TGF-β signals from the membrane to the nucleus to mediate gene transcription and VSMC differentiation. The balance between Smad-induced recruitment of corepressors or coactivators to TGF-β-responsive genes is associated with activation of HDAC or HAT (p300), which then alters histone acetylation. Transforming growth factor β induces histone hyperacetylation at the VSMC marker gene SM22 promoter through recruitment of HATs, Smad3, SRF, and myocardin, demonstrating a role for HATs and HDACs in TGF-β activation of VSMC differentiation.58
A proposed example of metabolic memory stored in the histone code of VSMCs is found in the dysregulation of histone H3 methylation, an epigenetic mark usually associated with transcriptional repression in type 2 diabetes.59 In VSMCs derived from type 2 diabetic db/db mice, levels of H3K9me3 (H3 lysine-9 trimethylation), as well as its HMT, are both reduced at the promoters of inflammatory genes. This loss of repressive histone marks, leading to increased inflammatory gene expression, is sustained in VSMCs from db/db mice cultured in vitro, suggesting persistence of metabolic memory. These results suggest that dysregulation in the histone code in VSMCs is a potential mechanism for increased and sustained inflammatory response in diabetic patients who continue to exhibit “metabolic memory” and vascular complications after glucose normalization.60
Influence of Cell-Cell and Cell-Matrix Interactions
Many differential VSMC functions are influenced by cell-cell and cell-matrix adhesion receptors that are altered during phenotypic modulation and during response to injury or disease. Cell-cell adhesion receptors include cadherins and gap junction connexins; cell-matrix interactions are dependent upon combinations of integrins, syndecans, and α-dystroglycan.11
Cell-Cell Adhesion Molecules: Cadherins and Gap Junction Connexins
After investment of VSMCs to the EC layer of nascent vessels, vascular stabilization, also known as maturation,61 is regulated by the sphingosine 1-phosphate (S1P) receptor S1P1, a GPCR on ECs. S1P1 activates the cell-cell adhesion molecule N-cadherin in ECs and induces formation of direct N-cadherin-based junctions between ECs and VSMCs required for vessel stabilization.61 To maintain VSMC quiescence within the vascular wall, cadherin-mediated cell-cell adherens-type junctions between VSMCs inhibit VSMC proliferation, possibly by inhibiting the transcriptional activity of β-catenin, a component of the Wnt signaling pathway, which interacts with the intracellular domain of cadherins.62 Inhibition of β-catenin or stabilization of cadherin junctions in VSMCs may be useful in treating vascular disease or injury.
Another type of direct intercellular junction between cells in the vasculature is the gap junction.63 Gap junctions, formed by connexin proteins between ECs and VSMCs and between VSMCs, are intercellular channels that allow movement of metabolites, small signaling molecules, and ions between cells.63–65 Of the four connexin proteins expressed in VSMCs (Cx37, Cx40, Cx43, and Cx45), Cx45 is exclusively found in VSMCs, while Cx43 is the most prominent and is essential for coordination of proliferation and migration.63 Homotypic gap junctions between VSMCs coordinate changes in membrane potential and intracellular Ca2 +, and heterotypic contacts between ECs and VSMCs at the myoendothelial junction control vascular tone by EC-mediated VSMC hyperpolarization. Notably, expression and/or activity of vascular connexins are altered in vascular diseases such as hypertension, atherosclerosis, or restenosis64 and in diabetes.63
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


