Chapter 6 Vascular Pharmacology
Vascular Smooth Muscle Activation
The actions of many drugs discussed in this chapter affect vascular smooth muscle cell (VSMC) contraction, and a basic understanding of contractile regulation is essential to understanding their mechanism of action. Contraction of vascular smooth muscle involves a sliding filament mechanism similar to that observed in other smooth muscle or in skeletal muscle. This topic has been reviewed in depth previously,1 is covered in detail in Chapter 3, and is therefore only briefly discussed here. The classical paradigm, depicted in Figure 6-1, is that increases in intracellular calcium lead to formation of a calcium-calmodulin complex. Calcium–CaM then binds and activates myosin light chain kinase (MLCK), which then phosphorylates myosin light chain (LC-20). Phosphorylation of LC-20 increases myosin adenosine triphosphatase (ATPase) activity, which leads to cross-bridge cycling and contraction. Myosin light chain phosphatase negatively regulates this process by dephosphorylating LC-20. Myosin light chain phosphatase is in turn inhibited by the small G-protein Rho and Rho kinase, which phosphorylates a subunit of myosin light chain phosphatase known as the myosin-binding subunit (MBS), leading to inhibition of phosphatase activity and favoring contraction. Myosin phosphatase is also inhibited by a 17-kDa protein known as CPI-17 (protein kinase C [PKC]–potentiated inhibitory protein of 17 kDa) that in turn is activated by PKC. Thus, activation of PKC can indirectly reduce myosin phosphatase activity, increase myosin phosphorylation, and promote vasoconstriction.
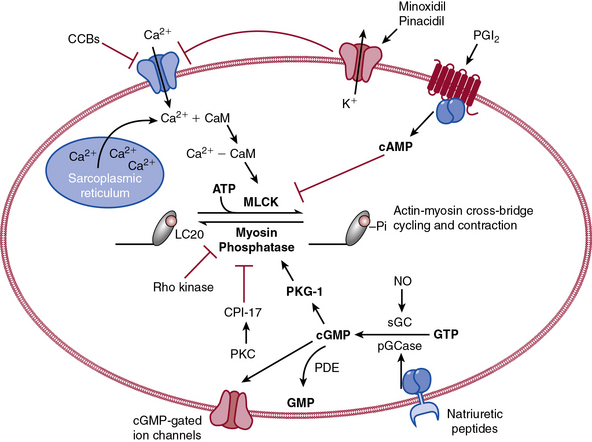
An important counterregulatory pathway in this scheme is the nitric oxide (NO) pathway. Nitric oxide acts on soluble guanylyl cyclase (sGC), which catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). In turn, cGMP acts as the only substrate for type 1 protein kinase G (PKG), which phosphorylates MBS, increasing its phosphatase activity and promoting vasodilation. Protein kinase G also phosphorylates and inhibits Rho, further reducing the propensity for vasoconstriction and promoting vasodilation. These pathways are targets of myriad vasoactive drugs that will be considered in greater depth in this chapter and are depicted in Figure 6-1.
It is now apparent that many pharmacological agents not only modulate vascular tone but also vascular growth, remodeling, inflammation, thrombosis, and vascular repair. As examples, many components of the contractile pathway discussed earlier exist in endothelial cells (ECs), including actin, myosin light chain, MLCK, Rho, and Rho kinase. These regulate endothelial shape, migration, cell-cell contact, and permeability. Myosin light chain kinase activation controls EC calcium entry, NO production, and release of endothelium-derived hyperpolarizing factor (EDHF).2 The Rho/Rho kinase pathway works in concert with other GTPases to modulate endothelial production of NO and reactive oxygen species (ROS) and gene expression.3 These aspects of vascular control have been the subject of substantial recent research, and new drugs have been developed to affect these targets. In addition, these pathways seem to be affected in an off-target fashion by several existing pharmacological agents.
Pharmacokinetics and Pharmacodynamics
The nature of a drug response helps classify the drug as a full or partial agonist, antagonist, or an inverse agonist (Fig. 6-2A) and may provide insight into the mechanism of drug action. For receptor conformation–specific drugs, pure antagonists stabilize the active and inactive conformations equally and have no net effect on basal activity. Inverse agonists preferentially stabilize the receptor’s inactive form, and agonists stabilize the active conformation.
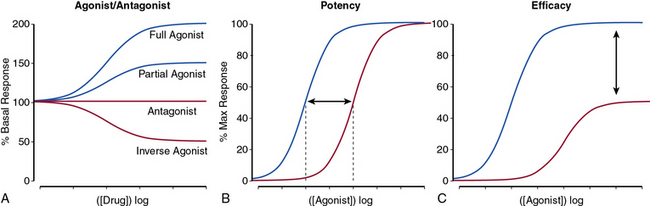
The potency of a drug refers to the molar concentration necessary to achieve a desired response (e.g., 50% maximal stimulation or inhibition; Fig. 6-2B), whereas efficacy reflects the drug’s maximal response relative to other agents (Fig. 6-2C). Clinical differences in drug potency may be overcome by increasing the dosage, whereas differences in drug efficacy cannot.
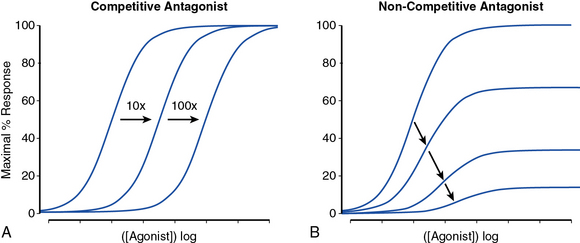
Receptor antagonists can be assessed by the response to a known stimulus in the presence of increasing antagonist concentration (Fig. 6-3). Antagonists that reversibly bind to the receptor can be overcome with increasing concentration of agonist (Fig. 6-3A). Antagonists that irreversibly bind their target impair the maximal response with increasing concentration (Fig. 6-3B). A number of drugs act in an allosteric manner by binding to a site on the receptor that is distinct from the native ligand, inducing a conformational change. Allosteric modulators can either increase or decrease agonist response by binding to a site distinct from the agonist binding site. An allosteric antagonist dose-response curve appears similar to that of a noncompetitive antagonist. Allosteric potentiators shift the agonist curve to the left (see Fig. 6-3A), while competitive antagonists shift the curve to the right.
Drugs That Affect Nitric Oxide/Guanylyl Cyclase/cGMP–Dependent Protein Kinase Pathway
The NO pathway plays a major role in modulating vascular reactivity; however, NO represents only one step in a complex pathway that can be affected by a variety of signaling molecules. This pathway is illustrated in the right portion of Figure 6-1, and involves the guanylyl cyclase enzymes, cGMP, and the binding targets of cGMP, which include the cGMP-dependent PKGs, ion channels regulated by cGMP, and phosphodiesterases (PDEs). The guanylyl cyclase/cGMP pathway is affected by a variety of agents, including NO and NO donors (the nitrovasodilators); other agents that activate guanylyl cyclase; agents that modulate degradation of cGMP; and agents that directly activate PKG.
Endogenously, NO is produced by the nitric oxide synthase (NOS) enzymes, and serves myriad signaling roles depending on the cell and tissue in which it is produced.4 Experimental studies have shown that NO produced by the endothelium not only mediates vasodilation, but also inhibits expression of adhesion molecules, reduces platelet adhesion, inhibits vascular smooth muscle growth and hypertrophy, and prevents vascular remodeling.
Nitrovasodilators
The nitrovasodilators produce their biological effects either by releasing NO or closely related molecules that are converted to NO in cells. The most commonly employed nitrovasodilators are the organic nitrates and sodium nitroprusside. It is useful to begin a discussion of these agents by comparing sodium nitroprusside and nitroglycerin, which are illustrated in Figure 6-4. As apparent, the oxidation state of the nitrogen that is ultimately released as NO differs in these molecules, and this basic structural property provides insight into their pharmacological profiles. Sodium nitroprusside requires a one-electron reduction to release NO, and this is readily accomplished nonenzymatically by a variety of reductants in the circulation, the interstitial space, and the cell. Thus, when infused intravenously, nitroprusside begins to release NO throughout the circulation and potently dilates all vessels. Moreover, given the short half-life of NO, the vasodilation caused by nitroprusside is short-lived once its infusion is discontinued.
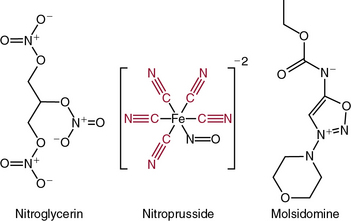
As is apparent from its structure, nitroprusside possesses five cyanide groups in each molecule (highlighted in red in Fig. 6-4), and prior studies have shown that each of these is reduced prior to the release of NO. The cyanide radicals react with hemoglobin (Hb) to form methemoglobin and are converted to thiocyanate in the liver. When these metabolic pathways are depleted, cyanide toxicity occurs, characterized by central nervous system (CNS) dysfunction, metabolic acidosis with a base deficit, and elevated plasma lactic acid concentrations.5 Fortunately, cyanide toxicity is infrequent during brief administration of sodium nitroprusside but occurs more commonly when infusion rates exceed 2 μg/kg/min and when the drug is infused for prolonged periods. In addition, the risk of cyanide toxicity is increased in patients with renal or hepatic failure, so sodium nitroprusside should be avoided in patients with these conditions. Owing to its capacity to rapidly release NO, sodium nitroprusside produces potent systemic vasodilation and is effective as an antihypertensive. It is still used for treatment of severe hypertension and, in some cases, for afterload reduction in patients with severe heart failure; however, newer agents with less potential toxicity are now more commonly used.
In contrast to sodium nitroprusside, nitroglycerin and other organic nitrates require a 3-electron reduction to yield NO. In the last several years, it has become clear that this is in large part accomplished by the action of the mitochondrial enzyme aldehyde reductase-2 (ADH2).6 Mice lacking this enzyme are resistant to the actions of nitroglycerin. Notably, about 40% of East Asians have a dominant negative mutation of ADH2 that causes intolerance to ethanol and markedly impaired vasodilator responses to nitroglycerin.7
As mentioned earlier, organic nitrates preferentially dilate larger arteries and veins while having less effect on arterioles, particularly at lower doses.8 This response profile is likely beneficial in alleviating angina because potent arteriolar dilators are prone to cause coronary steal and paradoxically worsen myocardial ischemia. Moreover, venous dilatation reduces left ventricular (LV) filling, alleviates pulmonary congestion, and can improve subendocardial perfusion in ischemic regions of the myocardium.
Experimental studies have shown that NO inhibits platelet adhesion, expression of adhesion molecules, and vascular smooth muscle proliferation and migration. Thus, one might expect that NO donors such as nitroglycerin would reduce atherosclerosis progression and potentially reduce major cardiovascular events in patients with coronary artery disease (CAD). Despite extensive use for alleviation of myocardial ischemia for almost a century and a half, no clinical trials have shown that these drugs reduce ischemic cardiovascular events. The GISSI-3 and ISIS-4 trials examined the effect of nitrates following myocardial infarction (MI) but failed to show a significant improvement in outcome.9,10 These trials only observed patients for 5 weeks to 6 months following MI, and therefore did not determine whether long-term nitrates might have a beneficial effect on outcome in patients with ischemic heart disease. Given the many putative beneficial effects of NO on vascular function, longer-term treatment might impart a beneficial effect on atherosclerosis, inflammation, vascular remodeling, or plaque stability. Indeed, a recent analysis of the GRACE registry, which includes patients admitted for ACS, showed that chronic nitrate users were much more likely to present with non–ST-segment elevation MI NSTEMI) than non-nitrate users.11 These data must be interpreted with caution because the use of nitrates was not randomized, and conclusions were derived from a retrospective analysis.
In addition to their use as antianginal agents, the long-acting nitrates are now often employed for treatment of congestive heart failure (CHF), commonly in combination with hydralazine. Unlike the case for treatment of CAD, prospective randomized trials have shown that long-acting nitrates improve survival, reduce hospitalizations, and enhance quality of life in patients with CHF, particularly among African Americans.12 Precise mechanisms underlying the beneficial effects are unclear; however, long- acting nitrates appear to synergize with hydralazine as afterload- and preload-reducing agents. These agents might also improve renal hemodynamics and promote diuresis and, via release of NO, have beneficial effects on vascular and cardiac remodeling.
A major limitation to prolonged use of organic nitrates is development of tolerance. Within about 12 hours of administration, the hemodynamic effects of organic nitrates begin to abate, in part due to extravascular adaptations such as volume redistribution and neurohormonal activation. After several days of continuous nitrate therapy, the direct vascular actions of nitrates are lost, even when vessels are removed from the animal or human. The mechanisms of nitrate tolerance, and in particular this latter form of true vascular tolerance, remain uncertain but have been attributed to formation of ROS, nitrosation and oxidation of guanylyl cyclase, and changes in activity of ADH2.13 A number of strategies have been proposed to prevent nitrate tolerance, but the only approach accepted clinically is to allow a drug “holiday”; that is, to withdraw the nitrate for about 12 hours daily. The commonly employed isosorbide mononitrate preparations accomplish this by increasing blood levels of the drug for about 12 hours during waking hours, after which blood levels fall to near-undetectable levels. Experimental studies have shown that hydralazine prevents nitrate tolerance by reducing oxidative stress,13 which might explain the benefit of hydralazine when added to long-acting nitrates in the treatment of heart failure. The long-acting nitrate pentaerythritol tetranitrate seems not to cause tolerance in experimental animals, but this has not been proven in clinical studies.
Intravenous nitroglycerin has occasionally been used for treatment of hypertensive emergencies. This condition is often associated with a contracted blood volume. Owing to nitroglycerin’s propensity to produce venular dilation rather than arteriolar dilation, it has the potential to reduce cardiac output in this setting and may produce untoward effects in patients with compromised coronary, renal, or cerebral perfusion.14 Low-dose nitroglycerin might be useful in combination with other agents in treating a hypertensive emergency, particularly in patients with acute pulmonary edema, but other agents are available and likely more effective.
In addition to its reaction with sGC, NO can react with other heme proteins and radicals. Higher oxides of NO can also react with thiols, leading to formation of nitrosothiols.15 An important example of these reactions is the reversible NO reaction with cytochrome C, which modulates mitochondrial respiration and superoxide production.16 It is uncertain as to how important these reactions are in the overall response to nitrovasodilators.
Related to the chemistry mentioned earlier are reactions of inorganic nitrate (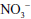 ) and nitrite (
) and nitrite (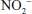 ). Although these are oxidation products of endogenously produced NO, they are also derived from dietary sources such as green leafy vegetables. Nitrate is rapidly converted to nitrite by bacteria in the oral cavity and gastrointestinal tract. Nitrite, in turn, can be reduced by various heme proteins, including deoxyhemoglobin, to NO. Studies have shown that the reaction of nitrite with deoxyhemoglobin promotes NO formation and vasodilation in regions of the circulation where oxygen tension is low, thereby improving oxygenation of hypoxic tissues.17 Thus, once considered an inactive metabolite of NO, nitrite likely has physiological significance and might have therapeutic utility.18
). Although these are oxidation products of endogenously produced NO, they are also derived from dietary sources such as green leafy vegetables. Nitrate is rapidly converted to nitrite by bacteria in the oral cavity and gastrointestinal tract. Nitrite, in turn, can be reduced by various heme proteins, including deoxyhemoglobin, to NO. Studies have shown that the reaction of nitrite with deoxyhemoglobin promotes NO formation and vasodilation in regions of the circulation where oxygen tension is low, thereby improving oxygenation of hypoxic tissues.17 Thus, once considered an inactive metabolite of NO, nitrite likely has physiological significance and might have therapeutic utility.18
Molsidomine (see Fig. 6-4) has also been used as an NO donor for treatment of angina, but it is not commonly employed clinically. The liver metabolizes molsidomine to release SIN-1, which in turn decomposes to NO and superoxide in equimolar amounts. These species can react rapidly with one another to yield the strong oxidant peroxynitrite. Because of this chemistry, SIN-1 oxidizes lipoproteins, damages DNA, and depletes antioxidants. This capacity to generate peroxynitrite has dampened enthusiasm for clinical use of molsidomine and related drugs, but SIN-1 is commonly used to produce peroxynitrite in experimental settings.
Unique Modulators of Soluble Guanylyl Cyclase
Compounds have been developed that activate sGC in an NO-independent fashion.19 Some of these, such as the pyrazolopyridine BAY 41-2272 and YC-1, interact with the heme group independent of NO, or can markedly enhance NO-stimulated enzyme activity. Others, such as BAY 58-2667 and HMR-1766, activate sGC in a heme-independent fashion and can stimulate cGMP formation even when the heme group is oxidized. Because these agents do not depend on endogenous production of NO, they have potential advantages over PDE inhibitors (see later discussion) in diseases where NO production is impaired. They also potentially bypass the problem of tolerance observed with various NO donors. These agents produce vasodilation, lower blood pressure, inhibit platelet aggregation, and have been shown to have therapeutic benefit in experimental models of systemic hypertension, pulmonary hypertension (PH), and heart failure.19 Like NO, they inhibit neointima formation following balloon injury in rats and therefore might be effective in treatment or prevention of restenosis and atherosclerosis. They also hold promise for treatment of erectile dysfunction (ED), liver fibrosis, and renal disease. Currently, clinical trials are underway to examine the efficacy of some of these agents in the treatment of heart failure and PH.
Natriuretic Peptides
Natriuretic peptides, including atrial (ANP), brain (BNP), and C-type (CNP) natriuretic peptides, are 17-amino-acid ring structures with an internal disulfide bond and are secreted as prohormones. Atrial natriuretic peptide and BNP are predominantly produced by atrial and ventricular myocytes; CNP is produced by vascular endothelial cells, the brain, and other peripheral tissues.20 Urodilatin, a related peptide processed from the ANP prohormone, is released from distal tubular cells of the kidney.21 The A and B natriuretic peptide receptors are homodimers that are widely distributed, particularly in the cardiovascular system and kidney.21 The cytoplasmic tails of these contain a guanylyl cyclase domain that is activated by binding with natriuretic peptides.20 There also exists a C-type natriuretic receptor that has a short cytoplasmic tail without a guanylyl cyclase domain and seems predominantly involved in clearing natriuretic peptides from the circulation.
As mentioned, ANP and BNP are produced predominantly in atrial myocytes. In the setting of a variety of conditions (e.g., heart failure, cardiac inflammation, fibrosis, hypoxia), BNP is expressed in large amounts by ventricular myocytes, leading to an elevation of circulating BNP. Thus, BNP and pro-BNP are commonly used as biomarkers for detection of various cardiac pathologies, and in particular for diagnosis and management of volume overload states.22
Activation of the A- and B-type natriuretic receptors leads to vasodilation and a variable diuretic and natriuretic response, depending on volume status. For this reason, a synthetic form of BNP known as nesiritide has been marketed and employed for treatment of decompensated heart failure. Like the nitrovasodilators, nesiritide infusion lowers pulmonary capillary wedge pressure (PCWP), right atrial pressure, and systemic vascular resistance, and improves symptoms of dyspnea.23 This agent also lowers circulating catecholamines, aldosterone, and angiotensin-(Ang) II levels, and aids diuresis. One study suggested that nesiritide was more effective that intravenous nitroglycerin treatment of patients with severe heart failure.23 An early meta-analyses suggested that nesiritide therapy was associated with an increase in mortality within 30 days of treatment, for uncertain reasons24; however, more recent meta-analysis of six randomized clinical trials showed no change in outcome at 10, 30, or 180 days following administration of this agent.25 A randomized trial of more than 7000 subjects has shown that treatment with nesiritide acutely improves patients with class IV heart failure, without worsening long-term outcome.26 This positive study is tempered by a very recent large study of 7143 patients with acute heart failure that showed no benefit of nesiritide in reducing symptoms or improving outcome at 30 days.27
Phosphodiesterase Inhibitors
As reflected in Figure 6-1, cGMP is rapidly inactivated to GMP by cellular PDEs. There are 11 PDE isoenzymes with varying specificities for the different cyclic nucleotides. Phosphodiesterases 5, 6, and 9 are highly selective for cGMP, while PDEs 3 and 10 are preferentially activated by cyclic adenosine monophosphate (cAMP). Phosphodiesterases 1, 2, and 11 have dual substrate specificity.28 In the cardiovascular system, the predominant PDEs are PDE1, 2, and 5. The PDEs are subject to substantial posttranslational regulation. As examples, PDE1 is calcium/CaM-dependent, cGMP stimulates PDE2 inactivation of cAMP, and binding of cAMP to PDE3 is inhibited by cGMP.
Several naturally occurring PDE inhibitors, such as caffeine, theophylline, and theobromines, are present in coffee, chocolates, and tea, and have been used since antiquity as stimulants.29 These are among the most widely distributed drugs in the world. Like cAMP and cGMP, the PDE inhibitors commonly contain a purine structure with linked pyrimidine and imidazole rings. These agents occupy the cAMP or cGMP PDE binding sites and inhibit respective PDE isoenzymes with varying degrees of selectivity.29 The immediate cardiovascular effects of nonselective PDE inhibition include vasodilation due to accumulation of cGMP and cAMP, increases in cardiac contractility due to accumulation of cAMP, and improvement in diastolic relaxation (lusitrophy) mediated by cAMP and cGMP.
In the past 30 years, a variety of PDE5 inhibitors, including sildenafil, tadalafil, and vardenafil, have been developed and are now used clinically (Table 6-1). Experimental studies have shown that the vasodilator effect of PDE5 inhibitors is almost exclusively dependent on endogenous NO release, and is prevented by inhibition of NOS and in conditions in which endogenous NO production is impaired.28 These agents also affect cardiac function. The PDE5 inhibitors acutely reduce cardiac contractility and precondition cardiac myocytes to reduce necrosis and apoptosis caused by experimental ischemia.30,31 Chronic PDE5 inhibition with sildenafil prevents experimental cardiac hypertrophy caused by transaortic constriction.32
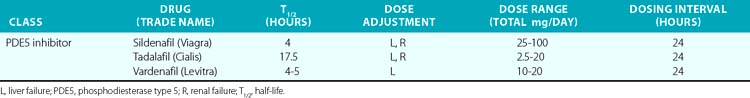
The PDE5 inhibitors were developed as antihypertensive agents, but because of their potent effect on the corpus cavernosa, they were initially approved and have become widely employed for treatment of erectile dysfunction. These agents are also potent dilators of the pulmonary circulation. Sildenafil and tadalafil been approved by the U.S. Food and Drug Administration (FDA) for treatment of pulmonary arterial hypertension (PAH). This disorder, defined by the hemodynamic parameters of a mean pulmonary artery pressure (PAP) above 25 mmHg and a PCWP 15 mmHg or lower, occurs as a primary condition and in the setting of a variety of diseases that affect the pulmonary circulation.33 (Also see Chapters 56 and 57.) A single dose of sildenafil was found to reduce PAP in patients with both primary and secondary PH and to augment the effect of inhaled NO in these subjects.34 Clinical studies have shown that chronic administration of PDE5 inhibitors reduces PAP and right ventricular (RV) mass, and improves exercise tolerance and functional status in patients with PAH.35 The recent SUPER-2 clinical trial showed that sildenafil improved 3-year survival in patients with PAH compared to historical controls.36 For these reasons, PDE5 inhibitors are now considered a mainstay of therapy for PAH. They have also been used with some success in neonates with persistent pulmonary hypertension of the newborn (PPHN).37
Phosphodiesterase type 5 inhibition has beneficial effects on hemodynamics and cardiac function in heart failure. In various experimental models of heart failure, PDE inhibitors prevent and reverse cardiac hypertrophy, reduce remodeling, and decrease myocardial fibrosis.38,39 In a recent placebo-controlled clinical trial of patients with severe heart failure, sildenafil treatment for 1 year improved ejection fraction, improved parameters of diastolic function, and reduced left atrial size while improving functional capacity and clinical status.40 This study was not designed to determine whether sildenafil improves survival; larger studies are needed with longer-term follow-up to discern whether PDE5 inhibition provides survival benefit. Nevertheless, these orally available agents, which avoid the problem of tolerance encountered with the nitrovasodilators, have substantial promise in treating ventricular dysfunction.
Prostaglandins and Thromboxane Agonists and Antagonists
Release of lipids from the cell membrane upon receptor binding or mechanical stimulation is a major signaling event in mammalian cells. One major class of lipid metabolites is the prostanoids, which include the prostaglandins (PGs) and thromboxane. The pathway leading to formation of these lipids is illustrated in Figure 6-5. They are formed from arachidonic acid, released from membrane phospholipids via the action of phospholipase A2. The initial step in prostanoid synthesis is conversion of arachidonic acid to the endoperoxide prostaglandin H2 (PGH2) by COX enzymes. Prostaglandin H2 is in turn a substrate for several enzymes including various PG synthases and thromboxane synthases (see Fig. 6-5), which leads to formation of multiple PG metabolites including PGE2, prostacyclin (PGI2), PGF2α, PGD2, and thromboxane A2 TxA2. Each of these has several G protein–linked receptors that are widely distributed and modulate myriad physiological and pathophysiological responses that include inflammation, vasomotor tone, hemostasis, renal function, and blood pressure.41,42 Vascular response to the various prostanoids depends on the category of the heterotrimeric G-protein receptor to which it binds. Vasodilator prostanoids, including PGI2 and PGD2, activate Gs, which leads to an increase in intracellular cAMP. The contractile prostanoids, including TxA2 and PGF2α, activate Gq, which leads to increased intracellular calcium. There are both Gs and Gq receptors for PGE2, which can therefore both vasodilate and vasoconstrict.
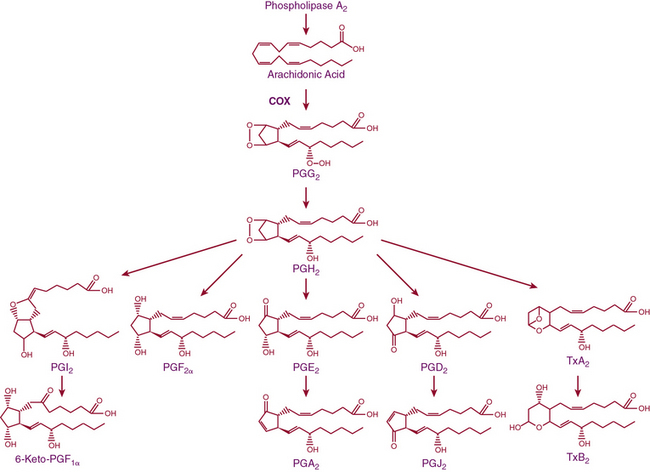
Figure 6-5 Arachidonic acid metabolic pathway.
COX, cyclooxygenase; PG, prostaglandin; PGI2, prostacyclin; Tx, thromboxane.
There are two isoforms of the COX enzymes: COX-1 and COX-2. Cyclooxygenase-1 is constitutively expressed and exerts housekeeping functions in many cells, including vascular cells. Cyclooxygenase-2 is generally considered an inducible enzyme, and its levels increase in the settings of inflammation, in particular when inflammatory cells enter the affected tissue.42 Cyclooxygenase-2 is also constitutively expressed in some cells, including ECs. The preferred substrate of COX-1 is arachidonic acid, but COX-2 can also produce unique antiinflammatory products from the endogenous cannabinoid 2-arachidonyl glycerol.43 Both COX-1 and COX-2 are activated by shear stress in the endothelium.44 The downstream products of COX are highly dependent on the cell type. In healthy blood vessels, the predominant arachidonic acid metabolite is PGI2, whereas platelets predominantly produce TxA2. In a variety of common cardiovascular diseases, however, vascular production of prostanoids can be shifted toward proinflammatory, procoagulant, and vasoconstrictor prostanoids.44 As an example, Ang-II stimulates COX-2 expression and production of PGE2 in VSMCs, and this response contributes to VSMC proliferation and migration in response to this hormone.45 In several experimental models of hypertension, obesity, and aging, the endothelium begins to produce prostanoid-contracting factors including PGH2, TxA2, and ROS generated as byproducts of COX activity.46
Cyclooxygenase Inhibitors
Aspirin has been studied extensively since the 1950s as a means of reducing cardiovascular events.47 Numerous large clinical trials performed in the 1980s supported the concept that aspirin decreases the occurrence of MI and stroke. A recent large meta-analysis showed that aspirin was effective in both primary and secondary prevention of total coronary events, ischemic stroke, and serious vascular events, with the greatest benefit observed in the case of secondary prevention.48 Another recent meta-analysis of nine trials that included 90,000 patients showed that aspirin is effective for primary prevention of nonfatal MI and total cardiovascular events, but not for stroke, cardiovascular mortality, or all- cause mortality.49 Of interest, several recent meta-analyses have suggested that aspirin might not be useful for primary prevention of events in the diabetic population.50,51
The beneficial effects of aspirin are generally considered a consequence of its antiplatelet effects and reduction of thrombosis. However, aspirin reduces levels of C-reactive protein (CRP) in patients with recent unstable coronary syndromes,52 and in experimental models of atherosclerosis, reduces atheroma burden, decreases inflammation, and improves endothelial function,53,54 suggesting that it might also have direct vascular effects.
Although aspirin has proven effective in reducing cardiovascular events, there are no clinical trials showing that other COX inhibitors convey similar cardiovascular benefit, and paradoxically, there is substantial evidence that these agents are harmful. The most striking example is that of the COX-2 inhibitor rofecoxib, which was withdrawn from the market because of increased thrombotic events55; however, other COX inhibitors might also increase cardiovascular risk, depending upon the relative COX-2–to–COX-1 selectivity.56,57 The precise mechanisms underlying this increased risk remain undefined, and it is unclear why aspirin, which inhibits the same enzyme, albeit via different mechanisms, is beneficial. These differences might relate to inhibition of vascular PGI2 and perhaps renal COX, which in turn could promote sodium retention and blood pressure elevation and worsen cardiovascular outcome. As previously mentioned, the downstream products and their receptors are myriad, so the in vivo actions of these agents are complex and difficult to predict. Nevertheless, NSAIDs other than aspirin should be used sparingly in patients with known cardiovascular diseases and currently have no role in preventing cardiovascular events.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


