Chapter 57 Pulmonary Hypertension in Non-Pulmonary Arterial Hypertension Patients
Overview of Pulmonary Hypertension
Definition and Nomenclature
Pulmonary hypertension (PH) is defined as a sustained mean pulmonary arterial blood pressure above 25 mmHg and pulmonary vascular resistance (PVR) of above 3 Wood units (240 dyne/s/cm− 5) at rest in the setting of a pulmonary capillary wedge pressure (PCWP) less than 15 mmHg.1 Pulmonary hypertension is a clinical syndrome typically characterized by hypoxemia, diminished exercise capacity, and dyspnea. In the vast majority of patients, PH develops as a consequence of hypoxic pulmonary vasoconstriction, vascular congestion, or impedance to pulmonary blood flow in the setting of primary lung, cardiac, or pulmonary vascular thromboembolic disease.
In contrast, mean pulmonary arterial hypertension (mPAH), a rare form of PH, results from an interplay between genetic and molecular factors that promotes pulmonary vascular endothelial dysfunction, vascular smooth muscle cell (VSMC) hypertrophy, and negative remodeling of small- and medium-sized pulmonary arteries in the absence of other cardiopulmonary disease.2 In PAH, pulmonary artery pressure (PAP) is often over 40 mmHg and may reach suprasystemic levels in severe cases; however, this occurs uncommonly in PH from non-PAH etiologies.3 Thus, PH and PAH are distinct pathophysiological entities, and although clinical signs and symptoms often overlap between these conditions, the terms are not synonymous.1
The contemporary PH classification system was created by an international panel of world experts at the 4th World Symposium on PH in 2008 (Dana Point, California) and broadly divides PH patients into two groups: those with PAH or PAH-associated conditions (i.e., formerly primary pulmonary hypertension) and PH that occurs in the setting of another cardiopulmonary disease (i.e., formerly secondary pulmonary hypertension).4
Chapter 56 is devoted to a discussion of PAH and PAH-associated conditions, whereas the current chapter provides an overview of the pathophysiology and treatment of disorders associated with secondary forms of PH. Specifically, this chapter will review primary diseases that modulate PH by causing (1) pulmonary venous hypertension, (2) chronic hypoxemia, (3) pulmonary thromboembolism, and (4) mechanical disruption to the normal pulmonary vasculature (i.e., World Health Organization [WHO] PH classification groups 2-5; Box 57-1).5 In clinical practice and throughout the published literature, the designation nonpulmonary arterial hypertension pulmonary hypertension is often invoked to describe these patients.
 Box 57-1 Clinical Classification of Pulmonary Hypertension by Etiology*
Box 57-1 Clinical Classification of Pulmonary Hypertension by Etiology*
1. Pulmonary arterial hypertension (PAH)
2. Pulmonary hypertension (PH) with left heart disease
3. PH associated with lung disease and/or hypoxemia
4. PH due to chronic thrombotic and/or embolic disease
5. Miscellaneous causes of PH (sarcoidosis, malignancy, fibrosing mediastinitis, Takayasu’s arteritis, iatrogenic)
Adapted from Simonneau G, Robbins IM, Beghetti M, et al: Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 54(1 Suppl):S43–S54, 2009.
Epidemiology, Diagnosis, and Natural History
The prevalence of PH in the general population is not well established, and PH incidence varies substantially according to primary disease subtype. A recent report of 455 patients with an elevated left ventricular (LV) end-diastolic pressure (but without left-sided valvular disease) observed that over half had comorbid PH,6 whereas PH is present in more than 90% of selected chronic obstructive pulmonary disease (COPD) patients.7,8 Rates of PH vary significantly within a specific primary disease subpopulation as well. For example, Handa et al. reported a 5% PH prevalence rate in one cohort of asymptomatic or mildly symptomatic sarcoidosis patients despite abnormal chest radiography, restrictive pattern on pulmonary function testing (PF T), and decreased peripheral oxygen saturation levels. If persistent dyspnea is present in sarcoidosis, however, PH prevalence rates increase to over 50%.9,10
The likelihood of developing clinically evident PH is often tightly linked to comorbid cardiac or lung disease characteristics. For example, symptomatic PH due to impaired LV diastolic function from chronic systemic hypertension is an indolent process that progresses with respect to decline in myocardial compliance.11 In contrast, severe PH from acute altitude sickness occurs via sudden hypoxia-mediated pulmonary microvascular vasoconstriction and hyperemia, which may occur independently of pulmonary reserve.12
Pulmonary hypertension prognosis, treatment choice, and clinical response to therapy are strongly associated with PH disease subtype. At present, goal-directed medical therapy for restoration of pulmonary microvascular function with calcium channel blockers, endothelin receptor antagonists (ERAs), inhaled nitric oxide (iNO), selective phosphodiesterase type 5 (PDE5) inhibition, or prostanoid replacement therapy is approved by the U.S. Food and Drug Administration (FDA) for use only in PAH patients. As is discussed in greater detail later, conclusions from small clinical trials have demonstrated a favorable effect of various PAH therapies on pulmonary hemodynamics and exercise tolerance in some WHO Class 2 thorough 5 conditions,13 but inappropriate administration of advanced PAH therapies to non-PAH patients with PH is likely to be ineffective and possibly harmful.1,6 Therefore, the emphasis of contemporary diagnostic algorithms is on discrimination of PAH from non-PAH patients1 (Fig. 57-1A). Comprehensive clinical, radiographic, serological, echocardiographic, and/or invasive hemodynamic testing is often necessary to confirm the absence of disease states that predispose to PH prior to making the diagnosis of PAH7 (Fig. 57-1B).
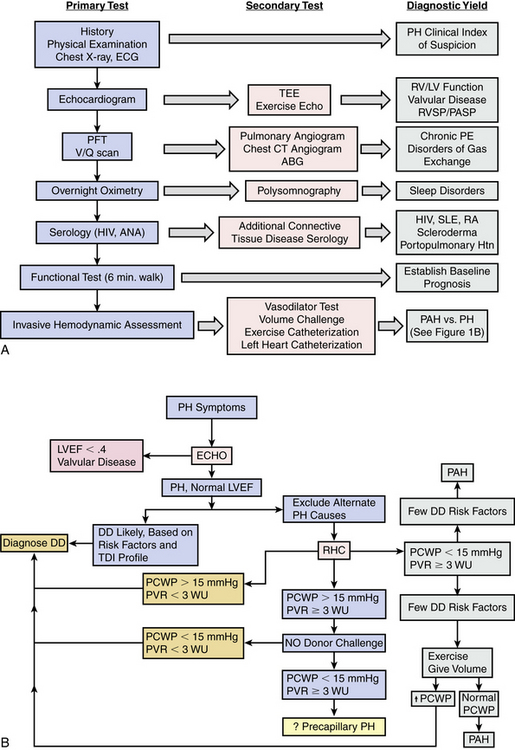
Figure 57-1 Pulmonary hypertension (PH) diagnostic algorithm.
(A adapted from McLaughlin VV, Archer SL, Badesch DB, et al: ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. J Am Coll Cardiol 53:1573–1619, 2009. B adapted from Hoeper MM, Barbera JA, Channick RN, et al: Diagnosis, assessment, and treatment of non-pulmonary arterial hypertension pulmonary hypertension. J Am Coll Cardiol 54[1 Suppl]:S85–S96, 2009.)
Pulmonary hypertension is underrecognized in clinical practice.14 Initiation of diagnostic testing for PH therefore requires a low index of clinical suspicion among practitioners who must recognize clues that suggest PH pathophysiology, such as familial or genetic risk factors for PAH, or comorbid conditions known to promote elevations in PAP.
Pulmonary Venous Hypertension
Pathophysiology of Pulmonary Hypertension due to Left-Sided Heart Disease
Acute left heart failure
Among the most common causes of PH is left-sided cardiac disease from LV systolic or diastolic dysfunction, or left-sided valvular disease (Table 57-1). In nonvalvular forms of left-sided heart disease, increased LV end-diastolic filling pressure is transmitted retrograde to the pulmonary venous and arterial circulatory beds. Acute changes to normal LV pressure-volume hemodynamics, such as occurs during an acute myocardial infarction (MI) or as a consequence of mitral valve leaflet rupture, predispose to sudden and dramatic increases in left atrial pressure and PAP15 (Fig. 57-2). Owing to the noncompacted, thin-walled architecture of the right ventricle (RV), acute pressure loading is poorly tolerated and results in RV systolic dysfunction, a major determinant of outcome in PH.16 Acute increases in PAP results in a congestive vasculopathy characterized by decreased pulmonary arteriolar compliance and loss of normal autoregulation of pulmonary vasomotor tone.17 These pathophysiological changes are generally reversible with pharmacotherapies that promote pulmonary vasodilation, reduce cardiac preload (e.g., nitric oxide [NO] donors), or directly alleviate pulmonary vascular congestion (e.g., loop diuretics).
Table 57-1 Cardiovascular Diseases That Predispose to Pulmonary Hypertension
| Clinical Feature | Mechanism |
|---|---|
| Chronic systemic hypertension | ↑ Left ventricular (LV) afterload → LV hypertrophy → ↓ LV compliance → ↑ LV end-diastolic filling pressure → pulmonary venous hypertension |
| Diabetes mellitus | Intramyocardial microcirculatory and epicardial coronary vascular dysfunction → LV systolic or diastolic dysfunction → pulmonary venous hypertension |
| Coronary artery disease (CAD) | Myocardial ischemia → LV systolic or diastolic dysfunction → pulmonary venous hypertension |
| Atrial fibrillation | Loss of “atrial kick” → ↑ left atrial and pulmonary venous congestion |
| Impaired diastolic function | ↑ End-diastolic filling pressure secondary to restrictive, infiltrative, or genetic cardiomyopathy → pulmonary venous hypertension |
| Mitral stenosis | ↑ Transmitral valve pressure ± atrial fibrillation → ↑ pulmonary venous hypertension |
| Mitral regurgitation (MR) | Chronic LV volume overload → LV cavitary dilation → ↑ LV end-diastolic filling pressure → pulmonary venous hypertension With elevated mitral regurgitant fraction, pulmonary artery pressure (PAP) is elevated secondary to pulmonary circulatory volume and pressure overload, particularly during exercise |
| Aortic insufficiency | Chronic LV volume overload → LV cavitary dilation → ↑ LV end-diastolic filling pressure → pulmonary venous hypertension |
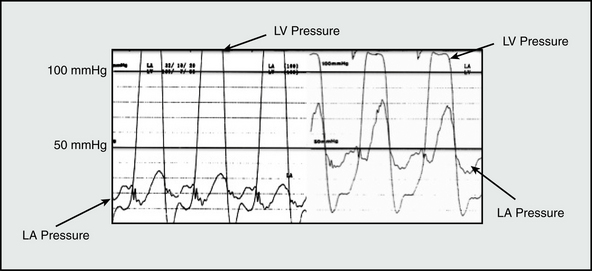
Chronic nonvalvular left-sided heart failure
In addition to passive pulmonary vascular congestion, circulating levels of the vasoactive peptide endothelin-1 (ET-1) positively correlate with PH severity in chronic left-sided heart failure.18 Pathophysiological concentrations of ET-1 disrupt normal vasomotor tone via activation of ETA/B receptors on VSMCs that increases intracellular [Ca+ 2]i levels. Endothelin-1 also promotes release of catecholamines (e.g., norepinephrine).19,20 Together, these processes are linked to VSMC contraction and in PH offset vasodilatory cell signaling pathways, resulting in pulmonary VSMC vasoconstriction.
Chronically elevated pulmonary venous pressure induces a cellular environment in pulmonary arterioles characterized by inflammation and increased levels of reactive oxygen species (ROS) generation. Over time, these maladaptive molecular processes are implicated in the development of irreversible pathological changes to normal pulmonary blood vessel architecture, including intimal fibrosis and VSMC hypertrophy and proliferation. Chronic RV pressure overload is also linked to the propagation of worsening left-heart failure by promoting abnormal changes in RV chamber deformation that adversely influences LV geometry.21
Diagnosis, Treatment, and Natural History
Pulmonary artery pressure, PVR, and RV systolic function are independent predictors of outcome in patients with chronic left-sided heart failure. In one study of 377 patients undergoing right heart catheterization with low LV ejection fraction and a history of congestive heart failure, mean pulmonary artery pressure (mPAP) over 29 mmHg portended about a threefold higher 36-month mortality rate (irrespective of RV function) compared to patients with normal mPAP. 22 Interpretation of pulmonary hemodynamics, however, must occur according to an individual patient’s specific clinical scenario. Although typically, PAP positively correlates with PH severity in chronic left-sided heart failure, this is not always the case. Since the generation of PAP is dependent on RV systolic function, abnormally low PAP may be observed in severe PH with RV failure. In this scenario, left-sided heart failure–mediated pulmonary vascular congestion results in an increased PVR even if PAP is normal or low.18 Pulmonary hemodynamic indices commonly used in clinical practice are provided in Table 57-2.
Table 57-2 Pulmonary Hemodynamic Measurements and Normal Ranges
| Measurement | Equation | Normal Range |
|---|---|---|
| Mean RA BP | Directly measured (PA catheter) | 0-8 mmHg |
| Pulmonary artery (PA) BP | Directly measured (PA catheter) | Systolic (PASP): 15-25 mmHg Diastolic (PADP): 4-12 mmHg mPAP 10-20 mmHg |
| PA capillary wedge pressure | Directly measured (PA catheter) PASP + (2 × PADP)]/3 | 6-12 mmHg |
| Cardiac output | Heart rate × Stroke volume/1000 | 4-7 L/min |
| PVR | 80 × (mPAP − PCWP)/cardiac output | 20-130 dyne/s/cm− 5 or 0.25-1.6 Wood units |
| Transpulmonary gradient | Mean SBP − Mean RABP | 5-8 mmHg |
BP, blood pressure; mPAP, mean pulmonary artery pressure; PA, pulmonary artery; PADP, pulmonary artery diastolic pressure; PASP, pulmonary artery systolic pressure; PCWP, pulmonary capillary wedge pressure; PVR, pulmonary vascular resistance; RA, right atrial; SBP, systolic blood pressure.
Conventional heart failure pharmacotherapy is the cornerstone treatment strategy for PH from nonvalvular left-sided cardiac disease. Angiotensin-converting enzyme (ACE) inhibitors, β-adrenergic receptor antagonists, loop diuretics, and vasodilators (e.g., hydralazine) often effectively decrease PVR and PAP, thereby promoting favorable responses in RV systolic function. A subset of chronic left-sided heart failure patients tends to exhibit a degree of PH that seems clinically and hemodynamically out of proportion to the severity of LV dysfunction, raising suspicion that alternative processes beyond passive vascular congestion alone are in play.1,7 Overall, sufficiently powered randomized clinical trials evaluating the effect of iNO, prostacyclin replacement, and ERAs for PH due to chronic left-sided heart failure have either failed to demonstrate a beneficial effect on pulmonary vascular hemodynamics or did so, but at the cost of significant adverse clinical events, including increased early mortality in one large trial of intravenous epoprostenol.23 Sildenafil, which promotes vasodilation by inhibiting PDE5 in lung VSMCs that consume cyclic guanosine monophosphate (cGMP), appears to decrease PAP and PVR safely without compromising cardiac output13 (Fig. 57-3). The effect of sildenafil on long-term outcome and survival in chronic heart failure patients is not yet known.
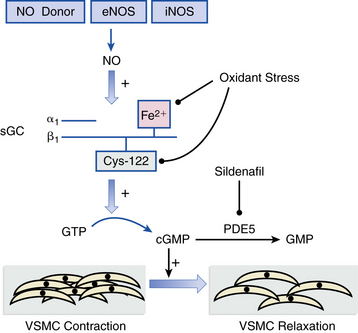
Pulmonary Hypertension due to Left-Sided Valvular Disease
Pulmonary hypertension due to left-sided valvular disease most often occurs as a consequence of mitral regurgitation (MR) or mitral stenosis and less commonly from severe aortic regurgitation. In aortic stenosis, initial pressure loading–induced LV hypertrophy is protective against PH. However, in decompensated aortic stenosis, LV cavitary dilation due to volume overload is associated with progressive PH.24 The final common pathway in the pathophysiology of PH, irrespective of valve lesion, is pulmonary venous hypertension. In only MR, however, PH is a key determinate for the timing of surgical valve therapy. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend mitral valve surgery (class IIa, level of evidence C) in asymptomatic patients with severe MR, preserved LV function, and PH (systolic PAP >50 mmHg).25 Magne et al. reported that in a cohort of 78 asymptomatic patients with at least moderate MR from degenerative mitral valve disease, resting PH (systolic PAP >60 mmHg) and exercise-induced PH were associated with a significantly lower 2-year symptom free survival rate (36 ± 14% vs. 59 ± 7% and 35 ± 8% vs. 75 ± 7%, respectively). Exercise-induced PH (particularly when systolic PAP >56 mmHg) was also an independent risk factor for development of symptoms.26 These data support exercise-induced PH as one potentially useful clinical marker for estimating the timing of surgical intervention for MR.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


