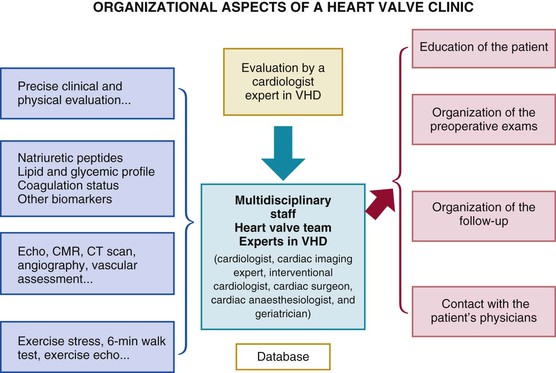
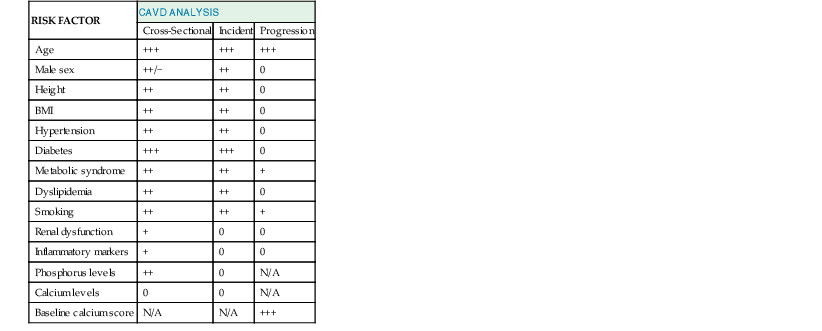
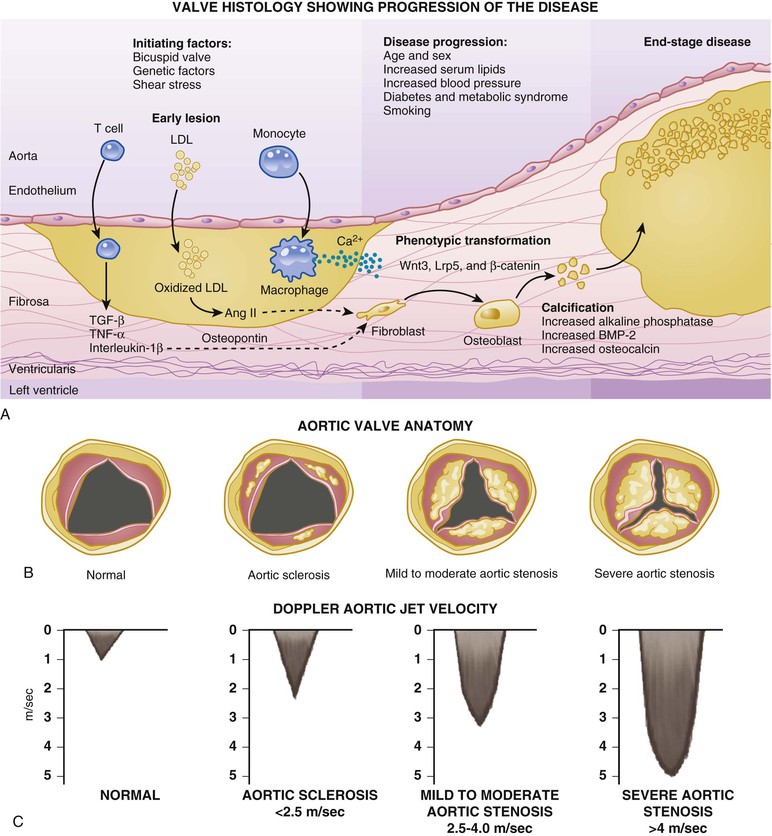
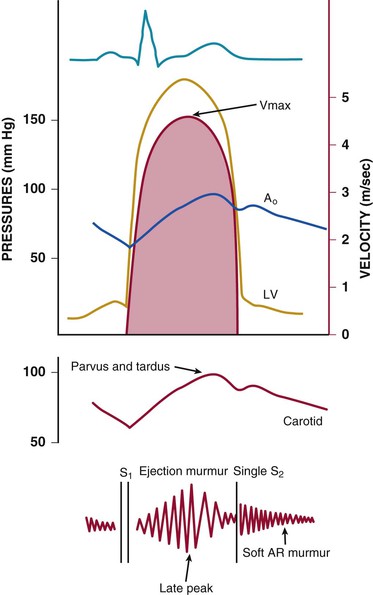
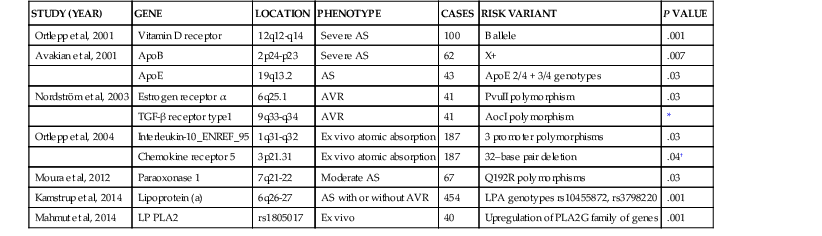
Catherine M. Otto, Robert O. Bonow Valvular heart disease accounts for 10% to 20% of all cardiac surgical procedures in the United States. The primary causes of valve disease are age-associated calcific valve changes and inherited or congenital conditions (e.g., a bicuspid aortic valve or myxomatous mitral valve disease). The prevalence of rheumatic valve disease now is very low in the United States and Europe because of primary prevention of rheumatic fever, although rheumatic valve disease remains prevalent in the developing world (see Chapter 83). Approximately two thirds of all heart valve operations are for aortic valve replacement (AVR), most often for aortic stenosis (AS). Mitral valve surgery most often is performed for mitral regurgitation (MR); most patients with mitral stenosis (MS) are treated by a percutaneous approach.1,2 In addition to patients with severe valve disease that eventually will require mechanical intervention, there is a larger group of patients with mild to moderate disease who need accurate diagnosis and appropriate medical management. Newer transcatheter techniques for treatment of valve dysfunction, including transcatheter aortic valve implantation (TAVI), device closure of paravalvular regurgitation, and several other methods (some investigational) of catheter-based mitral valve repair, are transforming our approach to the management of patients with valvular heart disease (see Chapter 56). In addition, increased understanding of the basic biologic mechanisms leading to valve dysfunction offers the hope of effective medical interventions to prevent or slow the disease process in the near future. Treatment paradigms are shifting toward a multidisciplinary approach to patient management, involving cardiologists (invasive and noninvasive), surgeons, and nurses with expertise in valvular heart disease. This trend has stimulated the development of multispecialty care teams working together within institutional heart valve centers (Fig. 63-1; see also Table e63-1 TABLE e63-1 Specific Aims of the Heart Valve Clinic From Lancellotti P, Rosenhek R, Pibarot P, et al: ESC Working Group on Valvular Heart Disease position paper. Heart valve clinics: Organization, structure, and experiences. Eur Heart J 34:1597, 2013. Valvular AS has three principal causes: a congenital bicuspid valve with superimposed calcification, calcification of a normal trileaflet valve, and rheumatic disease (Fig. 63-2). In a U.S. series of 933 patients undergoing AVR for AS, a bicuspid valve was present in more than 50%, including two thirds of those younger than 70 years and 40% of those older than 70 years of age.3 In addition, AS may be caused by a congenital valve stenosis manifesting in infancy or childhood. Rarely, AS is caused by severe atherosclerosis of the aorta and aortic valve; this form of AS occurs most frequently in patients with severe hypercholesterolemia and is observed in children with homozygous type II hyperlipoproteinemia. Rheumatoid involvement of the valve is a rare cause of AS and results in nodular thickening of the valve leaflets and involvement of the proximal portion of the aorta. Ochronosis with alkaptonuria is another rare cause of AS. Fixed obstruction to left ventricular (LV) outflow also may occur above the valve (supravalvular stenosis) or below the valve (discrete subvalvular stenosis; see Fig. 14-46) (Videos 63-1A and 63-1B Congenital Aortic Valve Disease. Congenital malformations of the aortic valve (see Chapter 62) may be unicuspid, bicuspid, or tricuspid, or the anomaly may manifest as a dome-shaped diaphragm (see Fig. 14-45). Unicuspid valves typically produce severe obstruction in infancy and are the most frequent malformations found in fatal valvular AS in children younger than 1 year but also may be seen in young adults with an anatomy that mimics bicuspid valve disease. Congenitally bicuspid valves rarely are responsible for serious narrowing of the aortic orifice during childhood4 but do cause significant aortic regurgitation (AR) requiring valve surgery in young adulthood in a subset of patients. Most affected patients, however, have normal valve function until late in life, when superimposed calcific changes result in valve obstruction (see later under Bicuspid Aortic Valve Disease). Calcific Aortic Valve Disease. Calcific aortic valve disease (formerly termed senile or degenerative) affecting a congenital bicuspid or normal trileaflet valve is now the most common cause of AS in adults. In a population-based echocardiographic study, 2% of persons 65 years of age or older had significant calcific AS (see Chapter 76), whereas 29% exhibited age-related aortic valve sclerosis without stenosis, defined as irregular thickening of the aortic valve leaflets detected by echocardiography without significant obstruction.5 Aortic sclerosis, identified by either echocardiography or computed tomography (CT), is the initial stage of calcific valve disease and, even in the absence of valve obstruction or known cardiovascular disease, is associated with a 50% increased risk of cardiovascular death and myocardial infarction over approximately 5 years of follow-up.6,7 Significant associations have been documented between calcific aortic valve disease and cardiovascular risk factors (Table 63-1). Although calcific AS once was considered to represent the result of years of normal mechanical stress on an otherwise normal valve, the evolving concept is that the disease process represents proliferative and inflammatory changes, with lipid accumulation, upregulation of angiotensin-converting enzyme (ACE) activity, increased oxidative stress, and infiltration of macrophages and T lymphocytes8–12 (Fig. 63-3) ultimately leading to bone formation13–15 in a manner similar, but not identical, to that for vascular calcification. Progressive calcification, initially occurring along the flexion lines at their bases, leads to immobilization of the cusps. A high prevalence of calcific AS also exists in patients with Paget disease of bone and end-stage renal disease. Age-related calcific AS shares common risk factors with mitral annular calcification, and the two conditions often coexist. Genetic polymorphisms have been linked to the presence of calcific AS, including those involving the vitamin D receptor, interleukin-10 alleles, and the apolipoprotein E4 allele (Table 63-2). Familial clustering of calcific AS also has been described, suggesting a possible genetic predisposition to valve calcification.16,17 The risk factors for the development of calcific AS are similar to those for vascular atherosclerosis—elevated serum levels of low-density lipoprotein (LDL) cholesterol and lipoprotein(a) [Lp(a)], diabetes, smoking, and hypertension.5,6,18 Calcific AS also has been linked to inflammatory markers and components of the metabolic syndrome. In a genome-wide association study based on a meta-analysis of data on nearly 7000 patients from three population-based cohorts, a single-nucleotide polymorphism in the locus for LDL was associated with aortic valve calcification, serum Lp(a) levels, and incident aortic stenosis (hazard ratio [HR], 1.68; CI, 1.32 to 2.15).19 This correlation was confirmed by review of a large Danish registry of more than 77,000 patients in whom two Lp(a) genotypes were significantly associated with incident AS.20 The growing consensus, therefore, is that “degenerative” calcific AS shares many pathophysiologic features with atherosclerosis and that specific pathways might be targeted to prevent or retard disease progression.5,11,13,21 TABLE 63-2 Candidate Gene Association Studies for Calcific Aortic Valve Disease * Significantly modified the effect of the estrogen receptor a polymorphisms (OR, 4.58; 95% CI, 1.68 to 12.51). † P value represents significance for effect modification with the interleukin-10 polymorphisms. Apo = apolipoprotein; LP PLA2 = lipoprotein-associated phospholipase A2. From Owens DS, O’Brien KD: Clinical and genetic risk factors for calcific valve disease. In Otto CM, Bonow RO (eds): Valvular Heart Disease: A Companion to Braunwald’s Heart Disease. 4th ed. Philadelphia, Saunders, 2013, pp 53-62. Data compiled from Ortlepp JR, Hoffmann R, Ohme F, et al: The vitamin D receptor genotype predisposes to the development of calcific aortic valve stenosis. Heart 85:635, 2001; Avakian SD, Annicchino-Bizzacchi JM, Grinberg M, et al: Apolipoproteins AI, B, and E polymorphisms in severe aortic valve stenosis. Clin Genet 60:381, 2001; Nordström P, Glader CA, Dahlén G, et al: Oestrogen receptor alpha gene polymorphism is related to aortic valve sclerosis in postmenopausal women. J Intern Med 254:140, 2003; Ortlepp JR, Schmitz F, Mevissen V, et al: The amount of calcium-deficient hexagonal hydroxyapatite in aortic valves is influenced by sex and associated with genetic polymorphisms in patients with severe calcific aortic stenosis. Eur Heart J 25:514, 2004; Moura LM, Faria S, Brito M, et al: Relationship of PON1 192 and 55 gene polymorphisms to calcific valvular aortic stenosis. Am J Cardiovasc Dis 2:123, 2012; Kamstrup PR, Tybjærg-Hansen A, Nordestgaard BG: Elevated lipoprotein(a) and risk of aortic valve stenosis in the general population. J Am Coll Cardiol 63:470, 2014; Mahmut A, Boulanger MC, El Husseini D, et al: Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: Implications for valve mineralization. J Am Coll Cardiol 63:460, 2014. Rheumatic Aortic Stenosis. Rheumatic AS results from adhesions and fusions of the commissures and cusps and vascularization of the leaflets of the valve ring, leading to retraction and stiffening of the free borders of the cusps. Calcific nodules develop on both surfaces, and the orifice is reduced to a small round or triangular opening (see Fig. 63-2C). As a consequence, the rheumatic valve often is regurgitant as well as stenotic. Patients with rheumatic AS invariably have rheumatic involvement of the mitral valve (see Chapter 83). With the decline in rheumatic fever in developed nations, rheumatic AS is decreasing in frequency, although it continues to be a major problem on a worldwide basis. In adults with calcific AS, a significant burden of leaflet disease is present before obstruction to outflow develops. However, once even mild obstruction is present, hemodynamic progression occurs in almost all patients, with the interval from mild to severe obstruction ranging from less than 5 to more than 10 years (Fig. 63-4). In infants and children with congenital AS, the valve orifice shows little change as the child grows, thereby contributing to the the relative obstruction over time. The clinical stages reflecting the progression of aortic stenosis are shown in Table 63-3. Severe obstruction to LV outflow usually is characterized by the following: (1) an aortic jet velocity of 4 m/sec or greater; (2) a mean systolic pressure gradient at least 40 mm Hg in the presence of a normal cardiac output; or (3) an effective aortic orifice (calculated by the continuity equation; see Fig. 14-49) no greater than 1.0 cm2 in an average-sized adult (i.e., <0.6 cm2/m2 of body surface area, approximately 25% of the normal aortic orifice of 3.0 to 4.0 cm2). An aortic valve orifice area between 1.0 to 1.5 cm2 is considered to represent moderate stenosis, and an orifice measuring greater than 1.5 to 2.0 cm2 is referred to as mild stenosis1,5,22,23 (see Table 63-3). The degree of stenosis associated with symptom onset varies among patients, however, and no single number defines severe or critical AS in an individual patient. Clinical decisions are based on consideration of symptom status and the LV response to chronic pressure overload, in conjunction with hemodynamic severity. In some cases, additional measures of hemodynamic severity, such as the energy loss index, valvular impedance, or evaluation with changing loading conditions (e.g., dobutamine stress) or with exercise, are necessary for full evaluation of disease severity.24–28 Chronic pressure overload typically results in concentric LV hypertrophy, with increased wall thickness and a normal chamber size. The increased wall thickness allows normalization of wall stress (afterload) so that LV contractile function is maintained. However, the increased myocardial cell mass and increased interstitial fibrosis result in diastolic dysfunction, which may persist even after relief of AS. Sex differences in the LV response to AS have been reported, with women more frequently exhibiting normal LV performance and a smaller, thicker-walled, concentrically hypertrophied left ventricle with diastolic dysfunction and normal or even subnormal systolic wall stress. Men more frequently demonstrate eccentric LV hypertrophy, excessive systolic wall stress, systolic dysfunction, and chamber dilation. The LV changes caused by chronic pressure overload are reflected in the LV and left atrial pressure waveforms and Doppler velocity curves. As contraction of the left ventricle becomes progressively more isometric, the LV pressure pulse exhibits a rounded, rather than flattened, summit, and the Doppler velocity curve exhibits a progressively later systolic peak. The elevated LV end-diastolic pressure and the corresponding Doppler changes in LV filling, which are characteristic of severe AS, reflect delayed relaxation and eventually decreased compliance of the hypertrophied LV wall. In patients with severe AS, large a waves usually appear in the left atrial pressure pulse and Doppler LV filling curve because of the combination of enhanced contraction of a hypertrophied left atrium and diminished LV compliance. Atrial contraction plays a particularly important role in filling of the left ventricle in AS: It raises LV end-diastolic pressure without causing a concomitant elevation of mean left atrial pressure. This “booster pump” function of the left atrium prevents the pulmonary venous and capillary pressures from rising to levels that would produce pulmonary congestion, while at the same time maintaining LV end-diastolic pressure at the elevated level necessary for effective contraction of the hypertrophied left ventricle. These changes in diastolic function are reflected in the Doppler parameter of LV filling and noninvasive measures of diastolic function, such as strain and strain rate (see Chapter 14). Loss of appropriately timed, vigorous atrial contraction, as occurs in atrial fibrillation (AF) or atrioventricular dissociation, may result in rapid clinical deterioration in patients with severe AS. Systemic vascular resistance also contributes to total LV afterload in adults with AS. Concurrent hypertension increases total LV load and may affect the evaluation of AS severity.29 Pulmonary hypertension is present in approximately 50% of adults undergoing AVR for severe AS and is associated with decreased long-term survival.30,31 The elevated pulmonary vascular resistance in AS patients decreases, with phosphodiesterase type 5 inhibition suggesting that the mechanism of pulmonary hypertension is increased LV preload and afterload.32 Exercise physiology is abnormal in adults with moderate to severe AS, and even asymptomatic patients have a reduced exercise tolerance. Although cardiac output at rest is within normal limits, the normal increase in cardiac output with exercise is blunted and is mediated primarily by increased heart rate, with little change in stroke volume. Even though stroke volume is unchanged, transvalvular flow rate increases because of the shortened systolic ejection period so that aortic jet velocity and transvalvular gradient increase proportionally. Before symptom onset, valve area increases slightly with exercise (by 0.2 cm2 on average), but as AS becomes more severe and symptoms are imminent, valve area becomes fixed, resulting in an even greater rise in jet velocity and pressure gradient with exercise. At this point, there is an abnormal blood pressure response to exercise (rise in systolic blood pressure <10 mm Hg), signifying severe valve obstruction. When the aorta is suddenly constricted in experimental animals, LV pressure rises, wall stress increases significantly, and both the extent and velocity of shortening decline. The development of LV hypertrophy is one of the principal mechanisms whereby the heart adapts to such an increased hemodynamic burden (see Chapter 21). The increased systolic wall stress induced by AS leads to parallel replication of sarcomeres and concentric hypertrophy. The increase in LV wall thickness is often sufficient to counterbalance the increased pressure so that peak systolic wall tension returns to normal, or remains normal, if the obstruction develops slowly. An inverse correlation between wall stress and ejection fraction has been described in patients with AS. This suggests that the depressed ejection fraction and velocity of fiber shortening that occur in some patients are a consequence of inadequate wall thickening, resulting in afterload mismatch. In others, the lower ejection fraction is secondary to a true depression of contractility; in this group, surgical treatment is less effective. Thus both increased afterload and altered contractility are operative to a variable extent in depressing LV performance. To evaluate myocardial function in patients with AS, ejection phase indices, such as ejection fraction and myocardial fiber shortening, should be related to the existing wall tension.33 Although LV hypertrophy is a key adaptive mechanism to the pressure load imposed by AS, it has an adverse pathophysiologic consequence (i.e., it increases diastolic stiffness; see Chapters 21 and 27). As a result, greater intracavitary pressure is required for LV filling. Some patients with AS exhibit an increase in stiffness of the left ventricle (increased chamber stiffness) simply because of increased muscle mass with no alteration in the diastolic properties of each unit of myocardium (normal muscle stiffness); others show increases in chamber and muscle stiffness. This increased stiffness, however produced, contributes to the elevation of LV diastolic filling pressure at any level of ventricular diastolic volume. Diastolic dysfunction may revert toward normal with regression of hypertrophy after surgical relief of AS, but some degree of long-term diastolic dysfunction typically persists. In patients with AS, coronary blood flow at rest is elevated in absolute terms but is normal when corrections are made for myocardial mass. Reduced coronary blood flow reserve may produce inadequate myocardial oxygenation in patients with severe AS, even in the absence of coronary artery disease. The hypertrophied LV muscle mass, increased systolic pressure, and prolongation of ejection all elevate myocardial oxygen consumption. The abnormally heightened pressure compressing the coronary arteries may exceed the coronary perfusion pressure and the shortening of diastole interferes with coronary blood flow, thus leading to an imbalance between myocardial oxygen supply and demand (see Fig. 63-4).34 Myocardial perfusion also is impaired by the relative decrease in myocardial capillary density as myocardial mass increases and by the elevation of LV end-diastolic pressure, which lowers the aortic-LV pressure gradient in diastole (i.e., the coronary perfusion pressure gradient). This underperfusion may be responsible for the development of subendocardial ischemia, especially when oxygen demand is increased or the diastolic filling period is reduced (e.g., with tachycardia, anemia, infection, or pregnancy). Clinical Presentation The cardinal manifestations of acquired AS are exertional dyspnea, angina, syncope, and ultimately heart failure.5,35,36 Most patients now are diagnosed before symptom onset on the basis of the finding of a systolic murmur on physical examination, with confirmation of the diagnosis by echocardiography. Symptoms typically begin at age 50 to 70 years with bicuspid aortic valve stenosis and in those older than 70 years with calcific stenosis of a trileaflet valve, although even in this age group approximately 40% of patients with AS have a congenital bicuspid valve.3 The most common clinical presentation in patients with a known diagnosis of AS who are followed prospectively is a gradual decrease in exercise tolerance, fatigue, or dyspnea on exertion. The mechanism of exertional dyspnea may be LV diastolic dysfunction, with an excessive rise in end-diastolic pressure leading to pulmonary congestion. Alternatively, exertional symptoms may be a result of the limited ability to increase cardiac output with exercise. More severe exertional dyspnea, with orthopnea, paroxysmal nocturnal dyspnea, and pulmonary edema, reflects various degrees of pulmonary venous hypertension. These are relatively late symptoms in patients with AS, and in current practice, intervention typically is undertaken before this disease stage. Angina occurs in approximately two thirds of patients with severe AS, approximately 50% of whom have associated significant coronary artery obstruction. It usually resembles the angina observed in patients with coronary artery disease (see Chapters 50 and 54) in that it is commonly precipitated by exertion and relieved by rest. In patients without coronary artery disease, angina results from the combination of the increased oxygen needs of hypertrophied myocardium and reduction of oxygen delivery secondary to the excessive compression of coronary vessels. In patients with coronary artery disease, angina is caused by a combination of epicardial coronary artery obstruction and the oxygen imbalance characteristic of AS. Very rarely, angina results from calcific emboli to the coronary vascular bed. Syncope most commonly is caused by the reduced cerebral perfusion that occurs during exertion when arterial pressure declines consequent to systemic vasodilation in the presence of a fixed cardiac output. Syncope also has been attributed to malfunction of the baroreceptor mechanism in severe AS (see Chapter 89), as well as to a vasodepressor response to a greatly elevated LV systolic pressure during exercise. Premonitory symptoms of syncope are common. Exertional hypotension also may be manifested as “graying-out spells” or dizziness on effort. Syncope at rest may be caused by transient AF with loss of the atrial contribution to LV filling, which causes a precipitous decline in cardiac output, or to transient atrioventricular block caused by extension of the calcification of the valve into the conduction system. Other late findings in patients with isolated AS include AF, pulmonary hypertension, and systemic venous hypertension. Although AS may be responsible for sudden death (see Chapter 39), this usually occurs in patients who had previously been symptomatic. Gastrointestinal bleeding may develop in patients with severe AS, often associated with angiodysplasia (most commonly of the right colon) or other vascular malformations. This complication arises from shear stress–induced platelet aggregation with a reduction in high-molecular-weight multimers of von Willebrand factor and increases in proteolytic subunit fragments.37 These abnormalities correlate with the severity of AS and are correctable by AVR. An increased risk of infective endocarditis has been documented in patients with aortic valve disease, particularly in younger patients with a bicuspid valve (see Chapter 64). Cerebral emboli resulting in stroke or transient ischemic attacks may be caused by microthrombi on thickened bicuspid valves. Calcific AS rarely may cause embolization of calcium to various organs, including the heart, kidneys, and brain. Physical Examination. The key features of the physical examination in patients with AS are palpation of the carotid upstroke, evaluation of the systolic murmur, assessment of splitting of the second heart sound (S2), and examination for signs of heart failure (see Chapter 11). The carotid upstroke directly reflects the arterial pressure waveform. The expected finding with severe AS is a slow-rising, late-peaking, low-amplitude carotid pulse, the parvus and tardus carotid impulse (Fig. 63-5). When present, this finding is specific for severe AS. However, many adults with AS have concurrent conditions, such as AR or systemic hypertension, that affect the arterial pressure curve and the carotid impulse. Thus an apparently normal carotid impulse is not reliable for excluding the diagnosis of severe AS. Similarly, blood pressure is not a helpful method for evaluation of AS severity. With severe AS, systolic blood pressure and pulse pressures may be reduced. However, in patients with associated AR or in older patients with an inelastic arterial bed, systolic and pulse pressures may be normal or even increased. Also with severe AS, radiation of the murmur to the carotids may result in a palpable thrill or carotid shudder. The cardiac impulse is sustained and becomes displaced inferiorly and laterally with LV failure. Presystolic distention of the left ventricle (i.e., a prominent precordial a wave) often is visible and palpable. A hyperdynamic left ventricle suggests concomitant AR and/or MR. A systolic thrill usually is best appreciated when the patient leans forward during full expiration. It is palpated most readily in the second right intercostal space or suprasternal notch and frequently is transmitted along the carotid arteries. A systolic thrill is specific, but not sensitive, for severe AS. Auscultation. The ejection systolic murmur of AS typically is late-peaking and heard best at the base of the heart, with radiation to the carotids (see Fig. 63-5). Cessation of the murmur before A2 is helpful in differentiation from a pansystolic mitral murmur. In patients with calcified aortic valves, the systolic murmur is loudest at the base of the heart, but high-frequency components may radiate to the apex—the so-called Gallavardin phenomenon, in which the murmur may be so prominent that it is mistaken for the murmur of MR. In general, a louder and later-peaking murmur indicates more severe stenosis. However, although a systolic murmur of grade 3 intensity or greater is relatively specific for severe AS, this finding is insensitive, and many patients with severe AS have only a grade 2 murmur. High-pitched decrescendo diastolic murmurs secondary to AR are common in many patients with dominant AS. Splitting of the second heart sound is helpful in excluding the diagnosis of severe AS because normal splitting implies the aortic valve leaflets are flexible enough to create an audible closing sound (A2). With severe AS, S2 may be single, because calcification and immobility of the aortic valve make A2 inaudible, closure of the pulmonic valve (P2) is buried in the prolonged aortic ejection murmur, or prolongation of LV systole makes A2 coincide with P2. Paradoxical splitting of S2, which suggests associated left bundle branch block or LV dysfunction, also may occur. Thus in older adults, normal splitting of S2 indicates a low likelihood of severe AS. The first heart sound (S1) is normal or soft, and a fourth heart sound (S4) is prominent, presumably because atrial contraction is vigorous and the mitral valve is partially closed during presystole. In young patients with congenital AS (see Chapter 62), the flexible valve may result in an accentuated A2 so that S2 may be normally split, even with severe valve obstruction. In addition, an aortic ejection sound may be audible because of the halting upward movement of the aortic valve. Like an audible A2, this sound is dependent on mobility of the valve cusps and disappears when they become severely calcified. Thus it is common in children and young adults with congenital AS but is rare in adults with acquired calcific AS and rigid valves. When the left ventricle fails and stroke volume falls, the systolic murmur of AS becomes softer; rarely, it disappears altogether. The slow rise in the arterial pulse is more difficult to recognize. Stated simply, with LV failure, the clinical picture changes from one of typical AS to that of severe LV failure with a low cardiac output. Thus occult AS may be a cause of intractable heart failure, and severe AS should be ruled out by echocardiography in patients with heart failure of unknown cause because operative treatment may be lifesaving and result in substantial clinical improvement. Dynamic Auscultation. The intensity of the systolic murmur varies from beat to beat when the duration of diastolic filling varies, as in AF or after a premature contraction. This characteristic is helpful in differentiating AS from MR, in which the murmur usually is unaffected. The murmur of valvular AS is augmented by squatting, which increases stroke volume. It is reduced in intensity during the strain of the Valsalva maneuver and on standing, both of which reduce transvalvular flow. Echocardiography (see Chapter 14) is the standard approach for evaluating and following patients with AS and selecting them for operation (see Figs. 14-47 to 14-50). Echocardiographic imaging allows accurate definition of valve anatomy, including the cause of AS and the severity of valve calcification, and sometimes allows direct imaging of the orifice area using three-dimensional imaging.22,38–40 Echocardiographic imaging also is invaluable for the evaluation of LV hypertrophy and systolic function, with calculation of ejection fraction, measurement of aortic sinus dimensions and detection of associated mitral valve disease.39 Doppler echocardiography allows measurement of transaortic jet velocity, which is the most useful measure for following disease severity and predicting clinical outcome. The stenotic orifice area is calculated using the continuity equation, and mean transaortic pressure gradient is calculated using the modified Bernoulli equation (see Fig. 14-49).39,40 Both valve area and pressure gradient calculations from Doppler data have been well validated compared with invasive hemodynamics and in terms of their ability to predict clinical outcome. However, the accuracy of these measures requires an experienced laboratory with meticulous attention to technical details. The combination of pulsed, continuous-wave and color flow Doppler echocardiography is helpful in detecting and determining the severity of AR (which coexists in approximately 75% of patients with predominant AS) and in estimating pulmonary artery pressure. In some patients, additional measures of AS severity may be necessary, such as correction for poststenotic pressure recovery or three-dimensional transesophageal imaging of valve anatomy (see Chapter 14). Evaluation of AS severity is affected by the presence of systemic hypertension so that reevaluation after blood pressure control may be necessary.41 In patients with LV dysfunction and low cardiac output, assessing the severity of AS can be enhanced by assessing hemodynamic changes during dobutamine infusion (see later). The severity of outflow tract obstruction gradually increases over 10 to 15 years, so the clinical course includes a long latent period during which stenosis severity is only mild to moderate and clinical outcomes are similar to those for age-matched normal patients.48,49 Of patients with mild valve thickening but no obstruction to outflow (e.g., aortic sclerosis), 16% will have valve obstruction at 1 year of follow-up, but only 2.5% will develop severe valve obstruction at an average of 8 years after the diagnosis of aortic sclerosis. Disease progression may be related to different factors than initiation of disease.50
Valvular Heart Disease
Overview
 ).
).

Aortic Valve Disease
Aortic Stenosis
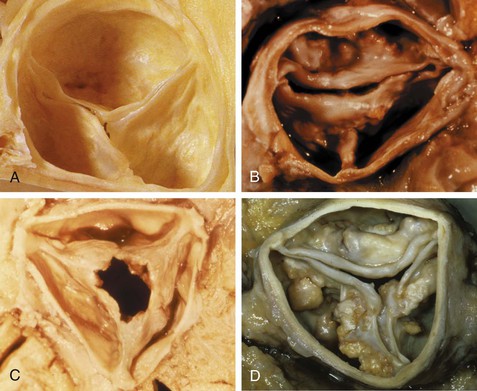
Causes and Pathology
 ); dynamic subaortic obstruction may be caused by hypertrophic cardiomyopathy (HCM) (see Chapter 66).
); dynamic subaortic obstruction may be caused by hypertrophic cardiomyopathy (HCM) (see Chapter 66).
STUDY (YEAR)
GENE
LOCATION
PHENOTYPE
CASES
RISK VARIANT
P VALUE
Ortlepp et al, 2001
Vitamin D receptor
12q12-q14
Severe AS
100
B allele
.001
Avakian et al, 2001
ApoB
2p24-p23
Severe AS
62
X+
.007
ApoE
19q13.2
AS
43
ApoE 2/4 + 3/4 genotypes
.03
Nordström et al, 2003
Estrogen receptor α
6q25.1
AVR
41
PvuII polymorphism
.03
TGF-β receptor type1
9q33-q34
AVR
41
AocI polymorphism
*
Ortlepp et al, 2004
Interleukin-10_ENREF_95
1q31-q32
Ex vivo atomic absorption
187
3 promoter polymorphisms
.03
Chemokine receptor 5
3p21.31
Ex vivo atomic absorption
187
32–base pair deletion
.04†
Moura et al, 2012
Paraoxonase 1
7q21-22
Moderate AS
67
Q192R polymorphisms
.03
Kamstrup et al, 2014
Lipoprotein (a)
6q26-27
AS with or without AVR
454
LPA genotypes rs10455872, rs3798220
.001
Mahmut et al, 2014
LP PLA2
rs1805017
Ex vivo
40
Upregulation of PLA2G family of genes
.001
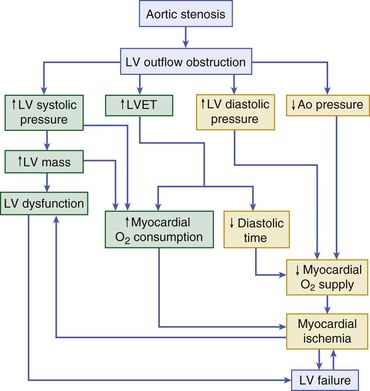
Pathophysiology
Myocardial Function in Aortic Stenosis
Diastolic Properties
Ischemia
Echocardiography
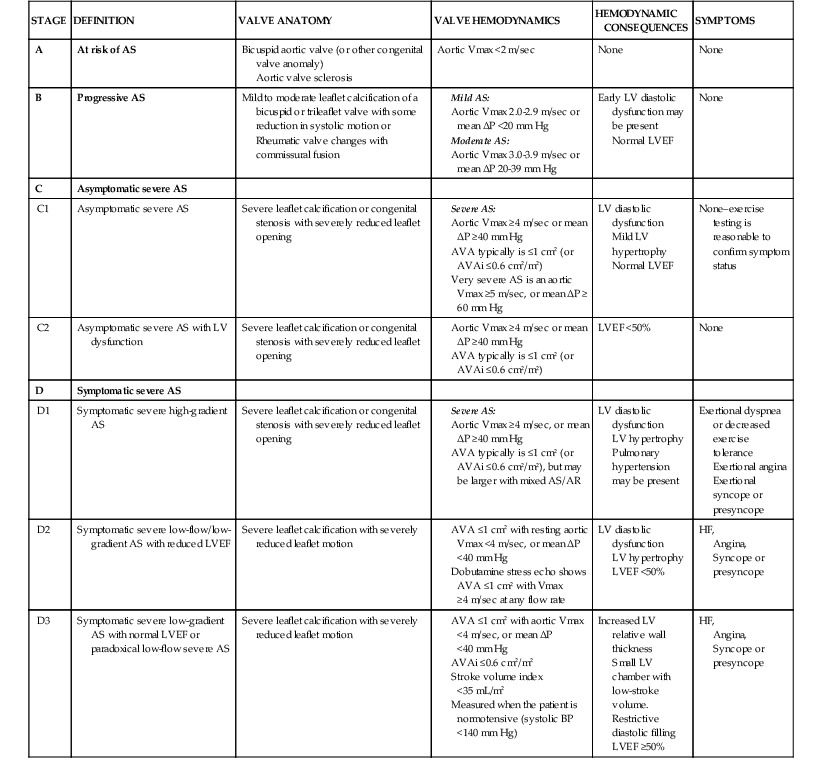
Disease Course
Clinical Outcome
Asymptomatic Patients.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Valvular Heart Disease
63