The therapeutic potential of vorapaxar in patients with non–ST-segment elevation acute coronary syndrome undergoing percutaneous coronary intervention (PCI) is unknown. This prespecified analysis of a postrandomization subgroup evaluated the effects of vorapaxar compared with placebo among Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRACER) participants undergoing PCI, focusing on the implanted stent type (drug-eluting stent [DES] vs bare-metal stent [BMS]). Among 12,944 recruited patients, 7,479 (57.8%) underwent PCI during index hospitalization, and 3,060 (40.9%) of those patients received exclusively BMS, whereas 4,015 (53.7%) received DES. The median (twenty-fifth, seventy-fifth percentiles) duration of thienopyridine therapy was 133 days (47, 246) with BMS and 221 days (88, 341) with DES. At 2 years among patients undergoing PCI, the primary (cardiovascular death, myocardial infarction, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization) and secondary (cardiovascular death, myocardial infarction, or stroke) end points did not differ between vorapaxar and placebo groups, which was consistent with the treatment effect observed in the overall study population (p value for interaction = 0.540). However, the treatment effect trended greater (p value for interaction = 0.069) and the risk for bleeding in patients taking vorapaxar versus placebo appeared attenuated in BMS-only recipients. After adjustment for confounders, the interaction was no longer significant (p value = 0.301). The covariate that mostly explained the stent-type-by-treatment interaction was the duration of clopidogrel therapy. In conclusion, among patients with PCI, the effect of vorapaxar is consistent with the overall TRACER results. Patients who received a BMS underwent shorter courses of clopidogrel therapy and displayed trends toward greater ischemic benefit from vorapaxar and lesser bleeding risk, compared with patients who received a DES.
In the large, phase III, Thrombin Receptor Antagonist for Clinical Event Reduction in Acute Coronary Syndrome (TRACER) trial, vorapaxar resulted in a nonsignificant relative reduction of 8% in the primary end point and in a nominally significant reduction of death from cardiovascular causes, myocardial infarction (MI), or stroke by 11% in 12,944 patients with non–ST-segment elevation acute coronary syndrome undergoing invasive or conservative management. A significant (58%) increase in Global Use of Strategies To Open Occluded Coronary Arteries moderate-to-severe bleeding complications was also noted. The combined inhibition of multiple platelet activation pathways has been shown to be particularly beneficial in patients undergoing coronary intervention with stent implantation. Hence, the objective of the present predefined subgroup analysis of the TRACER trial was to examine the efficacy and safety of vorapaxar compared with placebo in patients with non–ST-segment elevation acute coronary syndrome who underwent percutaneous coronary intervention (PCI) during the index hospitalization, with a focus on the type of implanted stent (drug-eluting stent [DES] vs bare-metal stent [BMS]).
Methods
The TRACER trial (funded by Merck & Co.; ClinicalTrials.gov number, NCT00527943 ) was an international, prospective, randomized, double-blind, event-driven trial in patients hospitalized for non–ST-segment elevation acute coronary syndrome managed invasively or medically. Patients were randomly assigned in a 1:1 ratio to receive vorapaxar (a loading dose of 40 mg and a daily maintenance dose of 2.5 mg thereafter) or matching placebo with stratification according to the intent to use a glycoprotein IIb/IIIa inhibitor (vs none) and the intent to use a parenteral direct thrombin inhibitor (vs other antithrombin agents). The recruitment period began on December 18, 2007, and ended on June 4, 2010. After a safety review in January 2011, the data and safety monitoring board recommended that the trial be terminated in a timely and orderly fashion, noting that the protocol-defined number of primary efficacy end points had been reached. Concerns regarding an increase in intracranial hemorrhages—when the trial had reached prespecified power on main efficacy analyses—was the main driver of the decision. Also, the board recommended stopping study medication in patients with a history of stroke who had been enrolled in a companion trial of patients with chronic vascular disease. The decision of whether to proceed to PCI and, if so, the type of stent (if any) to be implanted was left to the discretion of the treating physician.
To be eligible, patients had to experience symptoms of coronary ischemia within 24 hours before presentation and at least one of the following: cardiac troponin or creatine kinase-MB level higher than the upper limit of the normal, new ST-segment depression of >0.1 mV, or transient ST-segment elevation (<30 minutes) of >0.1 mV in at least 2 contiguous leads. Patients were also required to have one of the following: age of ≥55 years; previous MI, PCI, or coronary artery bypass graft surgery; diabetes mellitus; or peripheral artery disease. A complete list of inclusion and exclusion criteria has been published. The study protocol was approved by an independent ethics committee or institutional review board, and informed consent was required before any study procedure.
The study’s primary efficacy end point was the composite of cardiovascular death, MI, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization. The prespecified key secondary end point was the composite of cardiovascular death, MI, or stroke. Other efficacy end points are exploratory, including primary or key secondary end points in specific patient subsets, including patients who underwent PCI, or any analysis based on the type of implanted stent during the index procedure. End point definitions were previously reported.
The PCI population included patients who underwent PCI during the index hospitalization. Among those who had a stent implanted during the procedure, patients were further classified by the type of stent received. The BMS population includes patients who received only a BMS, and the DES population includes those who received at least 1 DES.
Baseline characteristics were compared between BMS and DES patients. Chi-square tests were used for categorical data, and Wilcoxon rank-sum tests were used for continuous variables. To analyze efficacy and bleeding end points after PCI, time-to-event analyses were performed. Event rates at 2 years were estimated by the Kaplan-Meier method. Hazard ratios (HRs) and 95% confidence intervals (CIs) were generated by Cox proportional hazards models. A propensity score of receiving a BMS or DES was estimated based on baseline characteristics and angiographic information ( Appendix 1 ). Inverse probability–weighted Cox models were fitted with the probability of receiving a BMS or DES represented by the propensity score. The same method was applied when comparing patients with and without PCI in terms of postdischarge bleeding events. The propensity of undergoing PCI was estimated and applied as the weight ( Appendix 2 ).
To investigate which factor(s) might have contributed to the differential vorapaxar effect on cardiovascular death, MI, or stroke among patients who received BMS and DES, we examined a list of characteristics that were clinically believed to be associated with both the end point and the choice for BMS or DES. The initial model included vorapaxar, DES, and the interaction between them. If (1) the vorapaxar-DES interaction went away after a characteristic (e.g., body weight) and its interaction with vorapaxar were added to the initial model and (2) the vorapaxar-body weight interaction became significant, then the characteristic (body weight) would be considered as a factor that explained the differential effect of vorapaxar among patients who received BMS and DES. Accordingly, a factor was considered to have explained the interaction if the following criteria were met: (1) by adding the interaction between the treatment and the tested covariate, the stent-type-by-treatment interaction becomes not significant and (2) the interaction between the treatment and the tested covariate is significant. To illustrate the effect of these effect modifiers, changes in p values of the vorapaxar-DES interaction were calculated when each characteristic was introduced to the initial model.
A 2-sided significance level of 0.05 was used for all tests. p Values were not adjusted for multiple comparisons. Because of the small amount of missing values in the analysis data sets, missing values were not imputed, and patients with missing values were excluded from analysis. SAS, version 9.2 (SAS Institute, Cary, North Carolina), was used for all analyses.
Results
A total of 12,944 patients at 818 sites in 37 countries were enrolled. Overall, 7,479 patients (57.8%) underwent PCI during index hospitalization, which did not differ between vorapaxar (3,764 of 6473 [58.1%]) and placebo (3,715 of 6,471 [57.4%]) groups ( Figure 1 ). Patients who were men, white, and recruited at European sites, with no history of MI, with stable hemodynamic conditions, and with greater prevalence of cardiac biomarker elevation or ST-segment deviation, yet with a less frequent history of renal insufficiency, were more likely to be treated with PCI in both treatment groups. Patients undergoing PCI were also more frequently treated with >100 mg/day of aspirin and concomitant thienopyridine therapy ( Supplementary Table ).
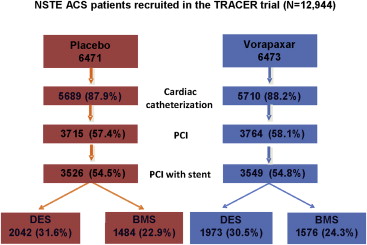
Among patients who underwent PCI, the procedure occurred within 24 hours of randomization in 2,982 (79.2%) of the 3,764 vorapaxar patients and in 2,929 (78.8%) of the 3,715 placebo patients. The first dose of study drug was given at a median of 3.5 hours before intervention in both study arms. As expected, stenting was accomplished in the vast majority of patients in both study groups (n = 3,549 [94.3%] in the vorapaxar group and n = 3,526 [94.9%] in the placebo group). Among patients with stents, at least 1 DES was implanted in 1,973 patients who received vorapaxar (55.6%) and in 2,042 patients who received placebo (57.9%), whereas 1,576 (44.4%) and 1,484 patients (42.1%) in the vorapaxar and placebo groups, respectively, received BMS only ( Figure 1 ).
BMS-treated patients were older and were less frequently diabetic, less often had a history of MI or coronary revascularization, and were less frequently recruited at North American sites; yet, they presented with a greater prevalence of ST-segment depression and a high Thrombolysis In Myocardial Infarction risk score. BMS-treated patients were also less likely to receive glycoprotein IIb/IIIa inhibitors, a >100 mg/day aspirin regimen, and concomitant thienopyridine therapy ( Table 1 ). Moreover, the median (twenty-fifth, seventy-fifth percentiles) duration of thienopyridine therapy was 133 days (47, 246) with BMS and 221 days (88, 341) with DESs (data not shown).
| Variable | PCI (N = 7479) | DES (N = 4015) | BMS (N = 3060) | p-Value DES vs. BMS |
|---|---|---|---|---|
| Age, years | <0.001 | |||
| Mean (SD) | 64.2 (10.0) | 63.6 (10.0) | 65.0 (9.7) | |
| Median (Q1, Q3) | 64.0 (57.0, 72.0) | 63.0 (57.0, 71.0) | 64.0 (58.0, 72.0) | |
| Range, min, max | 29.0, 93.0 | 29.0, 93.0 | 29.0, 92.0 | |
| Female | 1824/7479 (24.4%) | 1007/4015 (25.1%) | 732/3060 (23.9%) | 0.262 |
| White | 6475/7459 (86.8%) | 3410/4002 (85.2%) | 2730/3055 (89.4%) | <0.001 |
| Black | 143/7459 (1.9%) | 88/4002 (2.2%) | 43/3055 (1.4%) | |
| Asian | 637/7459 (8.5%) | 411/4002 (10.3%) | 175/3055 (5.7%) | |
| Other race | 204/7459 (2.7%) | 93/4002 (2.3%) | 107/3055 (3.5%) | |
| Body weight (kg) | <0.001 | |||
| Mean (SD) | 82.8 (17.5) | 83.6 (18.2) | 81.8 (16.7) | |
| Median (Q1, Q3) | 81.0 (70.5, 93.0) | 82.0 (71.0, 94.0) | 80.0 (70.0, 91.0) | |
| Range, minimum, maximum | 27.0, 181.0 | 39.5, 181.0 | 27.0, 176.9 | |
| <60 kg | 527/7455 (7.1%) | 285/3999 (7.1%) | 220/3054 (7.2%) | |
| Body mass index (kg/m 2 ) | 0.009 | |||
| Mean (SD) | 28.4 (5.1) | 28.6 (5.2) | 28.2 (4.9) | |
| Median (Q1, Q3) | 27.7 (25.1, 31.1) | 27.8 (25.2, 31.4) | 27.6 (25.0, 30.8) | |
| Range, minimum, maximum | 13.8, 63.1 | 15.2, 63.1 | 13.8, 62.6 | |
| North America | 1781/7479 (23.8%) | 1243/4015 (31.0%) | 458/3060 (15.0%) | <0.001 |
| Latin America | 358/7479 (4.8%) | 84/4015 (2.1%) | 262/3060 (8.6%) | |
| Western Europe | 3550/7479 (47.5%) | 1972/4015 (49.1%) | 1389/3060 (45.4%) | |
| Eastern Europe | 994/7479 (13.3%) | 224/4015 (5.6%) | 703/3060 (23.0%) | |
| Asia | 577/7479 (7.7%) | 369/4015 (9.2%) | 159/3060 (5.2%) | |
| Australia/New Zealand | 219/7479 (2.9%) | 123/4015 (3.1%) | 89/3060 (2.9%) | |
| Hypertension ∗ | 5184/7479 (69.3%) | 2783/4015 (69.3%) | 2094/3060 (68.4%) | 0.426 |
| Hyperlipidemia † | 4629/7476 (61.9%) | 2588/4013 (64.5%) | 1760/3059 (57.5%) | <0.001 |
| Diabetes mellitus | 2281/7478 (30.5%) | 1361/4014 (33.9%) | 771/3060 (25.2%) | <0.001 |
| Former smoker | 2225/7477 (29.8%) | 1187/4014 (29.6%) | 929/3059 (30.4%) | 0.158 |
| Current smoker | 2425/7477 (32.4%) | 1334/4014 (33.2%) | 968/3059 (31.6%) | 0.468 |
| Creatinine clearance (mL/min) | 0.147 | |||
| <30 | 77/7110 (1.1%) | 39/3788 (1.0%) | 29/2930 (1.0%) | |
| 30–60 | 798/7110 (11.2%) | 394/3788 (10.4%) | 349/2930 (11.9%) | |
| ≥60 | 6235/7110 (87.7%) | 3355/3788 (88.6%) | 2552/2930 (87.1%) | |
| Prior myocardial infarction | 2068/7479 (27.7%) | 1174/4015 (29.2%) | 724/3060 (23.7%) | <0.001 |
| Prior percutaneous coronary intervention | 1837/7476 (24.6%) | 1135/4013 (28.3%) | 524/3059 (17.1%) | <0.001 |
| Coronary bypass | 869/7479 (11.6%) | 506/4015 (12.6%) | 291/3060 (9.5%) | <0.001 |
| Stroke | 312/7479 (4.2%) | 161/4015 (4.0%) | 121/3060 (4.0%) | 0.905 |
| Peripheral artery disease | 548/7479 (7.3%) | 296/4015 (7.4%) | 212/3060 (6.9%) | 0.473 |
| Positive troponin or CK-MB | 7022/7426 (94.6%) | 3769/3991 (94.4%) | 2878/3033 (94.9%) | 0.405 |
| Electrocardiogram findings | ||||
| ST-segment depression | 2479/7479 (33.1%) | 1212/4015 (30.2%) | 1110/3060 (36.3%) | <0.001 |
| ST-segment elevation | 456/7479 (6.1%) | 245/4015 (6.1%) | 182/3060 (5.9%) | 0.787 |
| Symmetric T wave inversions | 1802/7479 (24.1%) | 869/4015 (21.6%) | 852/3060 (27.8%) | <0.001 |
| TIMI risk score | <0.001 | |||
| 0–2 | 36/7479 (0.5%) | 16/4015 (0.4%) | 18/3060 (0.6%) | |
| 3–4 | 3844/7479 (51.4%) | 2018/4015 (50.3%) | 1678/3060 (54.8%) | |
| 5–7 | 3599/7479 (48.1%) | 1981/4015 (49.3%) | 1364/3060 (44.6%) | |
| Killip class | 0.161 | |||
| I | 7130/7437 (95.9%) | 3847/3985 (96.5%) | 2917/3049 (95.7%) | |
| II | 244/7437 (3.3%) | 105/3985 (2.6%) | 108/3049 (3.5%) | |
| III–IV | 57/7437 (0.8%) | 30/3985 (0.8%) | 21/3049 (0.7%) | |
| Use or intent to use glycoprotein IIb/IIIa at randomization | 1614/7479 (21.6%) | 931/4015 (23.2%) | 612/3060 (20.0%) | 0.001 |
| Use or intent to use direct thrombin inhibitor at randomization | 1223/7479 (16.4%) | 723/4015 (18.0%) | 433/3060 (14.2%) | <0.001 |
| Thienopyridine at baseline | 6975/7479 (93.3%) | 3763/4015 (93.7%) | 2852/3060 (93.2%) | 0.379 |
| Aspirin dose at baseline (mg) | <0.001 | |||
| ≤100 | 4227/7254 (58.3%) | 2211/3896 (56.8%) | 1781/2966 (60.0%) | |
| 100–300 | 615/7254 (8.5%) | 316/3896 (8.1%) | 275/2966 (9.3%) | |
| ≥300 | 2412/7254 (33.3%) | 1369/3896 (35.1%) | 910/2966 (30.7%) |
∗ Defined as a history of hypertension as diagnosed by a physician or blood pressure of >140/90 mm Hg on at least 2 occasions. Current use of medical therapy to treat hypertension.
† Defined as a history of dyslipidemia diagnosed and/or treated by a physician or documentation of cholesterol >200 mg/dl, a low-density lipoprotein level of >130 mg/dl, a high-density lipoprotein level of <40 mg/dl.
At 2 years after the index PCI, the primary end point (cardiovascular death, MI, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization) occurred in 15.6% of patients who received vorapaxar and 16.7% of patients who received placebo (adjusted HR 0.96, 95% CI 0.84 to 1.09). The key secondary end point (cardiovascular death, MI, or stroke) occurred in 10.6% of patients who received vorapaxar and 12.5% of patients who received placebo (adjusted HR 0.90, 95% CI 0.78 to 1.05). These findings were consistent with the treatment effect observed in the overall study population. There was no significant interaction between randomized treatment and index PCI on primary and key secondary end points after index discharge (p = 0.540 for interaction on the primary end point, p = 0.555 for interaction on the key secondary end point).
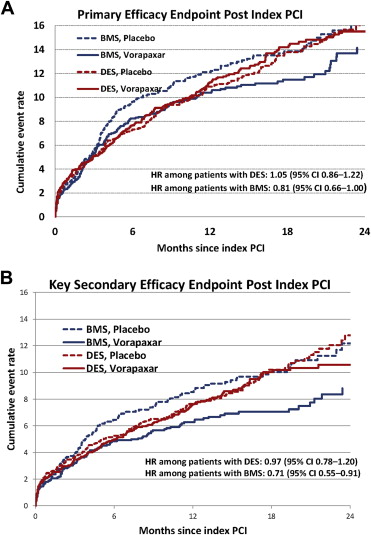
The treatment effect on the primary efficacy end point in patients who were treated exclusively with BMS or with at least 1 DES at the time of the index PCI procedure is shown in Figure 2 and Table 2 . At 2 years, the primary end point in BMS-only–treated patients trended lower in the vorapaxar group (14.4%) versus the placebo group (16.6%; adjusted HR 0.81, 95% CI 0.66 to 1.00). In patients who received DES, the 2-year event rate was 15.7% in the vorapaxar group and 16.3% in the placebo group (adjusted HR 1.05, 95% CI 0.86 to 1.22). There was no interaction between the type of stent and vorapaxar (adjusted p value = 0.175).
| Endpoint | Total Pts | Pts With Events, 2-Year KM % | HR (95% CI ∗ ) | Interaction p-Value (Treatment and Stent † ) | |
|---|---|---|---|---|---|
| Vorapaxar | Placebo | ||||
| Primary efficacy | 0.175 | ||||
| Drug-eluting stent | 4015 | 249 (15.7%) | 253 (16.3%) | 1.05 (0.86–1.22) | |
| Bare-metal stent | 3060 | 181 (14.4%) | 196 (16.6%) | 0.81 (0.66–1.00) | |
| Key efficacy secondary | 0.301 | ||||
| Drug-eluting stent | 4015 | 173 (10.6%) | 186 (12.7%) | 0.97 (0.78–1.20) | |
| Bare-metal stent | 3060 | 115 (9.3%) | 141 (12.0%) | 0.71 (0.55–0.91) | |
| Cardiovascular death | 0.470 | ||||
| Drug-eluting stent | 4015 | 41 (2.6%) | 37 (2.5%) | 1.28 (0.81–2.02) | |
| Bare-metal stent | 3060 | 37 (2.9%) | 40 (3.8%) | 0.88 (0.56–1.39) | |
| Myocardial infarction | 0.382 | ||||
| Drug-eluting stent | 4015 | 128 (8.1%) | 146 (9.9%) | 0.90 (0.70–1.14) | |
| Bare-metal stent | 3060 | 85 (7.0%) | 101 (8.2%) | 0.73 (0.54–0.98) | |
| Stroke | 0.851 | ||||
| Drug-eluting stent | 4015 | 20 (1.6%) | 23 (1.5%) | 0.94 (0.51–1.73) | |
| Bare-metal stent | 3060 | 14 (1.4%) | 18 (2.3%) | 0.69 (0.34–1.38) | |
| Definite stent thrombosis | 0.471 | ||||
| Drug-eluting stent | 4015 | 29 (1.6%) | 23 (1.3%) | 1.33 (0.76–2.31) | |
| Bare-metal stent | 3060 | 21 (1.4%) | 23 (1.6%) | 0.88 (0.48–1.61) | |
| Definite/probable stent thrombosis | 0.090 | ||||
| Drug-eluting stent | 4015 | 34 (1.8%) | 23 (1.3%) | 1.56 (0.91–2.67) | |
| Bare-metal stent | 3060 | 26 (1.7%) | 29 (2.0%) | 0.89 (0.52–1.51) | |
∗ Adjusted for baseline covariates and whether there was an event before PCI. The baseline covariates are listed in Appendix 1 .
† Accounted for propensity of receiving DES or BMS. Baseline covariates and angiographic findings for estimating the propensity score are listed in Appendix 2 .
The treatment effect on the key secondary end point trended greater in patients who were treated exclusively with BMS at the time of the index PCI procedure ( Figure 2 and Table 2 ). At 2 years, the key secondary end point in BMS-only–treated patients occurred in 9.3% of patients who received vorapaxar versus 12.0% of patients who received placebo (adjusted HR 0.71, 95% CI 0.55 to 0.91). In patients who received DES, the 2-year event rate was 10.6% in patients who received vorapaxar and 12.7% in patients who received placebo (adjusted HR 0.97, 95% CI 0.78 to 1.20). There was a noted trend for interaction between the type of stent and vorapaxar (p value = 0.069). However, after adjusting for baseline and angiographic characteristics as well as for concomitant medications, the interaction was no longer significant (p value = 0.301).
In the effect modifier analyses, the single covariate that most reduced the stent-type-by-treatment interaction factor was duration of clopidogrel therapy, entered as a time-dependent covariate ( Figure 3 ; interaction treatment × clopidogrel duration p = 0.207).



