Chapter 31 Tumor Immunology and Immunotherapy
Glossary of Terms
ADCC—antibody-dependent cellular cytotoxicity
CDC—complement-dependent cytotoxicity
CDR—complementarity-determining region
CpG—unmethylated CG dinudeotides
CTLA-4 cytotoxic T lymphocyte-associated antigen-4
EGFR—epidermal growth factor receptor
FDA—U.S. Food and Drug Administration
GM-CSF granulocyte macrophage-colony stimulating factor
HIV—human immunodeficiency virus
hTERT—human telomerase reverse transcriptase
IDO—indoleamine 2,3-dioxygenase
LAK—lymphokine-activated killer cell
MDSC—myeloid-derived suppressor cell
MHC—major histocompatibility complex
PARP—poly (ADP-ribose) polymerase
PD-L1—programmed death receptor ligand-1
PRR—pattern recognition receptor
PSMA—prostate-specific membrane antigen
SEREX—serologic expressive cloning
SOCS—suppressor of cytokine signaling
TGF-β—transforming growth factor β
TRAIL—TNF-related apoptosis-inducing ligand
VEGF—vascular endothelial growth factor
The immune system is our most powerful defense against infectious disease1 and the mediator of rejection in transplantation. In a small subset of patients, modern immune-based therapies can also effect dramatic and durable rejection of bulky metastatic melanoma and renal cell cancer. This attests to the capacity of the adaptive, and perhaps innate, immune system to recognize and destroy malignancies. These complete responses, however few, are durable, and patients rarely relapse, which is even more remarkable. Perhaps the most important observation, which has been confirmed repeatedly over the last 2 decades, is that the human immune system recognizes self-antigens expressed in the context of major histocompatibility complex (MHCs) on the cell surface. Almost half of the human T cell repertoire recognizes self—these generally low-affinity self-reactive T cells having escaped thymic deletion—which underscores the reality that antitumor immune responses are frequently autoimmune in nature. The immune system has evolved mechanisms of immunologic tolerance which allow for discrimination between self and nonself. Therefore, there is a wealth of potential immune targets for solid and hematopoietic malignancies, which include normal self-proteins and tumor-specific mutations, against which human T cells can be activated, expanded, and become specifically cytotoxic. Genome-based scientific advances have provided promising recombinant immunomodulatory molecules as well.2 Thus, there is broad potential in human cancer immunotherapy, if we could only understand all the biologic rules.
Overview of Tumor Immunology
T Lymphocytes and Natural Killer Cells
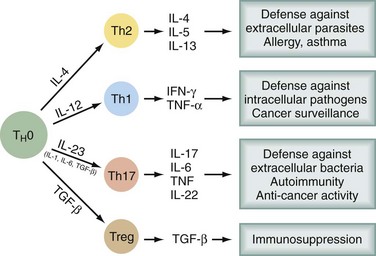
Bone marrow–derived progenitor cells enter the thymus, from which T cells eventually emerge. In the thymus, an enormous repertoire of T cell receptors is randomly generated by recombinations and mutations in α and β chains of the T cell receptor (TCR). Progenitors with TCRs of high affinity for self-antigens undergo deletion (negative selection). Some of those with low affinity for self-antigens survive and are positively selected, so that a significant percentage of self-reactive T cells emerge from the thymus. Only a very small percentage of the cells entering and proliferating within the thymus survive this education process. Several types of T cells emerge into the periphery. CD8+ T cells recognize antigen in the context of MHC class I molecules, express αβ TCR, have cytotoxic activity, and produce cytokines. CD4+ T cells recognize antigen in the context of MHC class II molecules. There are several subsets of CD4+ T cells (Fig. 31-1). Among the better recognized are Th1 cells (helper type 1 T cells) that secrete interleukin-2 (IL-2), tumor necrosis factor α (TNF-α), and interferon-γ (IFN-γ) and Th2 cells, which produce IL-4, IL-5, IL-6, IL-10, and IL-13. Th1 cells promote cytotoxicity and inflammation whereas Th2 cells assist in the stimulation of B cells to produce antibody. T helper cells will favor a Th1 (cell-mediated) or Th2 (humoral) immune response, but a subset of regulatory cells (Treg cells) plays a critical role in dampening autoimmunity. These Treg constitute 5% to 10% of CD4+ cells and express the transcription factor Foxp3. Mutation of the Foxp3 gene in humans and mice leads to multiorgan autoimmune disease. A fourth type of T cell subtype is the so-called Th17 cell, which preferentially produces IL-17, IL-21, and IL-22 and is important in the pathogenesis of autoimmune diseases.
CD4+ T cells also play an important role in the initiation and maintenance of CD8+ T cell responses.3 They may do this through a variety of mechanisms. Activated CD4+ T cells can interact with dendritic cells (DCs), professional antigen-presenting cells, through an interaction between the CD40 receptor and its ligand, CD40L. This activation, or licensing, of DCs allows these antigen-presenting cells to promote the differentiation of CD8+ T cells and establish a durable memory T cell response. CD4+ T cells also produce IL-2 and IFN-γ, which could potentially support CD8 function. Thus, the importance of CD4+ T cells in shaping a productive antitumor response has been incorporated into many tumor immunotherapy strategies.
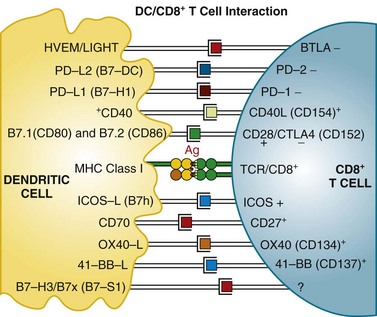
Mature T cells have a broad repertoire of αβ TCR, with diverse antigen specificity. This diverse TCR repertoire is generated during T cell differentiation by a process of gene rearrangement of variable (V), joining (J), and diversity (D) gene segments. TCRs are composed of α and β chains; it is estimated that recombination events could potentially yield a repertoire exceeding 1012 unique TCRs. These TCRs recognize antigen in the context of MHC proteins found on the surface of cells. Proteins within the cell are digested in the proteasome complex into short peptide fragments (8- to 12-amino-acid residues), which are transported to the cell surface bound in the groove of MHC class molecules; the specific peptide sequence presented is determined by the MHC (in humans, also called HLA [human leukocyte antigen]) allele. These class I–restricted peptides are recognized by CD8+ T cells. This provides the immune system with a continuous surveillance system for intracellular pathogens, such as viruses, so that infected cells can be quickly recognized and killed. The activation of resting T cells requires engagement of the correct MHC-peptide complex by the TCR (so-called signal 1) and additional costimulatory signals (signal 2). Professional antigen-presenting cells (DCs) provide the molecule B7 (either CD80 or CD86), which engages the CD28 receptor on the T cell, a requirement for T cell activation. T cells then upregulate another receptor, cytotoxic T lymphocyte–associated antigen 4 (CTLA-4), which also binds B7 but with a higher affinity than CD28. Engagement of CTLA-4 induces an inhibitory signal that downregulates T cell activation. This is a natural immunomodulatory mechanism for dampening immune responses. Monoclonal antibodies that bind to CTLA-4 can block this interaction and inhibit the negative regulatory signaling. Studies in human subjects have demonstrated that CTLA-4 blockade can break peripheral tolerance to self-antigens and induce antitumor and antiself autoimmune responses. Other examples of potential signaling interactions between DCs and T cells are shown in Figure 31-2 (see later).
Although much emphasis in antitumor immunity has been focused on adaptive responses (T lymphocytes and antibodies), effector cells of the innate immune system, specifically NK cells, can act alone or in concert with adaptive immunity.2,4 NK cells are innate immune cells because they can recognize and kill target cells without prior sensitization. These cells contain activating and inhibitory cell surface receptors and, when their activating receptors are engaged without concomitant ligation of their inhibitory receptors, can kill targets directly. NK cells have been traditionally viewed as providing a first line of defense, attacking virally infected cells. NK cells can also interact with the adaptive immune system. They can modulate the function of professional antigen-presenting cells (e.g., DCs), promote the generation of Th1 responses, and potentially dampen autoimmune immunopathology. Because their inhibitory receptors engage MHC molecules, NK cells specifically recognize cells that have lost MHC class I molecules; this can occur during viral infections or malignant transformation. NK cells are strongly activated by exogenous cytokines such as interleukin 2 (IL-2) and are then termed lymphokine-activated killer cells (LAK cells). LAK cells have greatly enhanced cytotoxicity for a much broader range of target cells.
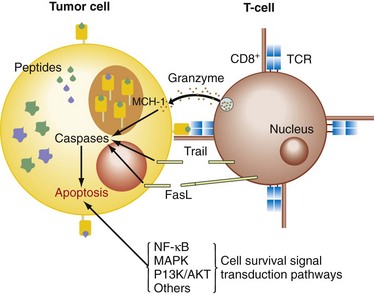
The cytotoxic pathways initiated by the activation of CTLs (cytotoxic T lymphocytes) and NK cells are the granule-exocytosis pathway, releasing membrane-destructive perforin and granzymes, and the death receptor pathways mediated by TNF-α, TRAIL, and FasL on binding their cognate receptors on the target cell surface (Fig. 31-3).5 These receptors activate the caspase cascade involving caspase-8 autoprocessing, activation of caspase-3, and cleavage of death substrates (e.g., poly[ADP-ribose] polymerase, PARP), with subsequent induction of apoptosis. Another mechanism inducing apoptosis, which is more typical for some chemotherapeutic drugs, uses a mitochondrial pathway involving permeabilization of the mitochondrial membrane and mitochondrial collapse. Mitochondrial integrity is preserved by a balance between antiapoptotic (e.g., Bcl-2, Bcl-xL, Bfl-1/A1, Mcl-1) and proapoptotic Bcl-2 family members (e.g., Bax, Bid, Bad, Bik, Bcl-xS). Mitochondrial destabilization will facilitate the cytosolic release of apoptogenic molecules, which expedites caspase-9 activation and in turn activates caspase-3, leading to apoptosis. The expression of these gene products is tightly regulated by the activity of survival signaling pathways (nuclear factor κB [NF-κB], AKT/PI3K, ERK1/2, p38, and JNK).
Antigen-Presenting Cells
DCs are professional antigen-presenting cells; their role is to take up, process, and present antigen to the immune system,6 and they are essential during the initial activation of resting T cells. There are different subtypes of DCs, with specialized functions that depend on their anatomic location. DCs are found in lymphoid tissues, in the skin, and on the mucosal surfaces of many organs. DCs in the gastrointestinal tract can sample bacteria in the intestinal lumen and initiate secretory immunoglobulin A (IgA) responses. In the lung, DCs help maintain tolerance to inhaled allergens. In the peripheral blood, DC precursors can migrate to sites of inflammation and initiate immune responses. DC function is powerfully modulated by a variety of receptors including Toll-like receptors (TLRs) and surface C-type lectin receptors. DCs at different stages in differentiation vary in their ability to migrate, take up antigen by phagocytosis, and effectively stimulate T cells. Immature DCs patrol their environment, sampling by pinocytosis and receptor-mediated endocytosis. Extracellular antigens are taken up into endosomes, which fuse with protease-containing lysosomes.
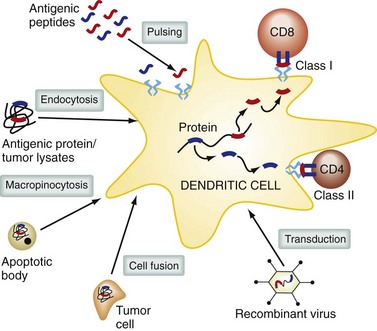
Within these compartments, antigens are cleaved into peptides, which can bind to MHC class II molecules and be delivered to the cell surface. Proteins in the cytoplasmic compartment of antigen-presenting cells are degraded by the proteosome and actively transported into the endoplasmic reticulum, where they are loaded onto MHC class I molecules and delivered to the cell surface. Some exogenous or environmental antigens can also find their way into the MHC class I antigen presentation pathway; this is termed cross-presentation and is an important mechanism for generating CD8+ class I–restricted T cell responses (Fig. 31-4). DCs can acquire antigen in the periphery and travel to lymph nodes, where they interact with T cells and present antigen. DCs originate from pluripotent stem cells from bone marrow, enter the blood, and localize to almost all tissues and lymphoid organs. There are a number of different subsets of DCs, including myeloid DCs (these include DCs found in deep epithelial tissues and Langerhans cells present in the epidermis) and plasmacytoid DCs, which are a major source of type I IFN.
Tumor Antigens
A molecular understanding of tumor recognition has been achieved only recently. The first molecularly defined antigen recognized by a tumor-reactive T cell was only discovered in 1991.7 This advance first required elucidation of the biology of antigen processing and presentation and its interaction with MHC molecules, which occurred in the late 1980s. These discoveries showed that when antigens were presented by MHC class I molecules, they were largely derived from intrinsic cytoplasmic proteins. These were degraded (predominantly in the proteosome) to specific 8- to 12-amino-acid peptides, which were actively transported into the endoplasmic reticulum, where they were loaded onto nascent MHC class I molecules, specifically binding to the antigen groove of a particular presenting MHC allele. These MHC-peptide complexes are then exported to the cell surface and are the actual entities that engage a specific TCR. Antigens presented by class II MHC molecules are often environmental proteins taken up by professional antigen-presenting cells (DCs) by endocytosis; these endosomes fuse with lysosomes, which mediate degradation of those exogenous proteins into short amino acid fragments that are loaded onto class II molecules and again displayed on the cell surface as peptide-MHC complexes. One cannot discern from its sequence or structure whether a particular TCR recognizes a class I– or class II– restricted antigen. Mature T cells also express the CD8 or CD4 coreceptor and these bind to invariant portions on all class I or class II MHC molecules, respectively. This additional ligation increases the affinity of the T cell interaction with the antigen-presenting cell. Therefore, T cells expressing CD4 typically recognize antigens presented by MHC class II molecules and CD8+ T cells usually recognize class I–presented antigens.
A comprehensive analysis of human cancer antigens has identified 46 that are immunogenic in clinical trials, with 20 having suggestive therapeutic efficacy.8 Examples of general classes of tumor antigens include the following: (1) MAGE-1, -2, and -3, BAGE, and RAGE, which are nonmutated antigens expressed in a variety of tumor cells; (2) lineage-specific tumor antigens, such as the melanocyte-melanoma lineage antigens MART-1/Melan-A (MART-1), gp100 protein, mda 7 protein, tyrosinase, and tyrosinase-related-protein (TRP-1 and -2), and the prostate antigens—prostate-specific membrane antigen (PSMA) and prostate-specific antigen (PSA); (3) epitopes derived from genes mutated in tumor cells and/or genes expressed at different levels in tumor compared with normal cells, such as mutated ras, bcr/abl rearrangement, and p53; (4) epitopes derived from oncoviral processes, such as human papillomavirus (HPV), and proteins E6 and E7; and (5) nonmutated proteins with a tumor-selective expression, including carcinoembryonic antigen (CEA), PSA, Her2/neu, and α-fetoprotein, among a rapidly growing list. Although the immune system has been widely exposed to some of these in fetal life or later, responses can still be generated to these proteins when adequately presented to the immune system (Box 31-1).
Box 31-1 Tumor Antigens Recognized by T Cells
Immunosuppressive Tumor Microenvironment
There is abundant evidence that cancer cells have acquired an array of defense mechanisms to thwart destruction by the immune system.9 These are summarized in Box 31-2. Most human cancers present peptide epitopes in the context of MHC molecules that can be recognized by antigen-reactive T cells, but tumor cells themselves do not present antigen in an immunostimulatory context. The human T cell repertoire that recognizes tumor self-antigens generally have TCRs of low affinity and require additional signaling through costimulatory molecules, such as B7-1/B7-2 (CD80-CD86) for optimal T cell activation and expansion. Without these other signals, T cells can become anergic. Tumor cells can also downregulate antigen expression by a variety of mechanisms, such as epigenetic silencing, loss of MHC expression, and loss of function of the intracellular machinery that processes and transports peptides to the cell surface. These antigen loss variants and lack of costimulatory signaling impose limitations on tumor cells, autonomously initiating antitumor immune responses.
Box 31-2 Cancer Cell Defense Mechanisms
The immune system also has complex and generally fine-tuned downregulatory signaling so that immune responses can be appropriately modulated.10 Shutting down an acute immune response after 1 to 2 weeks may be appropriate for a viral infection, but can be an impediment to rejecting a large mass of malignant tissue. Many autoimmune processes and transplant rejection, which can serve to model tumor rejection, are chronic ongoing events and tumor immunotherapy will require the circumvention of the normal protective mechanisms that prevent these occurrences.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


