TRUNCUS ARTERIOSUS
Introduction
Truncus arteriosus (TA) is a conotruncal anomaly characterized by origin of a common arterial trunk from the heart, almost always overriding a ventricular septal defect (VSD) and the common arterial trunk gives rise to coronary arteries, aorta (Ao) and pulmonary arteries (PAs) (Figure 32.1).1 Several different names have been used in the literature to describe this entity and these include, TA communis, TA communis persistens, common aorticopulmonary trunk, persistent TA, persistent arterial trunk and others. TA accounts for approximately 1% to 2% of congenital heart defects (CHDs).2,3 In this chapter, anatomy, classification, clinical presentation, physiology and management of TA in the neonate will be discussed. The readers interested in issues beyond the neonatal period are referred to comprehensive review published elsewhere.4
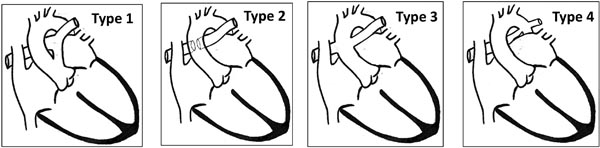
Figure 32.1. The Collett and Edwards classification of truncus arteriosus10 based on origin of PAs. Type 4 in this classification is no longer considered TA, but is considered a type of pulmonary atresia with VSD.
Pathology and Classification
In TA, both great arteries arise as one common arterial trunk. The PAs arise either as a main pulmonary artery (MPA), which then bifurcates, or as individual branch PAs originating directly from the ascending portion of common arterial trunk without an MPA. VSD is of conal septal type and is almost always present. VSD may extend into the membranous area of the ventricular septum. VSD typically creates an appearance of overriding of the common trunk in a parasternal long-axis view on echocardiogram, similar to that seen in tetralogy of Fallot (TOF), as explained in the imaging section. Nature of branching pattern of PAs from the common trunk forms the basis of the widely used Collett and Edwards classification described below.
Associated lesions are present in 21% of lesions5–9 and include right aortic arch (16–40%), interrupted aortic arch (10%), atrial septal defect (10%), persistent left superior vena cava (SVC) (6%), mitral valve anomalies (10%), coronary osteal stenosis, absent right pulmonary artery (RPA) or left pulmonary artery (LPA), among others.
Of the several classifications that exist, Collett and Edwards10 is commonly used (Figure 32.1) and is based on the nature of origin of PAs.
Type 1: A short segment of MPA is present, usually originating from left-posterior location. MPA divides into RPAs and LPAs.
Type 2: MPA is absent. Both branch PAs arise close to each other, from the posterior part of the common trunk.
Type 3: MPA is absent. Both branch PAs arise from the respective sides of the common trunk.
Type 4: MPA is absent. Both branch PAs arise from descending aorta (DAo). This type, also called “pseudotruncus,” is no longer considered a type of TA, but as a type of pulmonary atresia with VSD.
Embryology
TA is thought to result from failure of septation of the conotruncus portion of the heart tube during embryonic development. Several theories have been put forth, including failure of migration of neural crest cells to the region of branchial arches leading to failure of formation of spiral septum that separates the primitive common arterial trunk (TA) into Ao and PA.11 As the spiral septum contributes to formation of the conus portion of the ventricular septum, there is almost always a VSD in the conal septal area.
Genetics
Patients with DiGeorge syndrome have a high incidence of conotruncal anomalies. About 20% to 33% of patients with TA have 22q11 microdeletion. At the present time, DiGeorge syndrome, velocardiofacial syndrome, and chromosome 22q11 microdeletion are considered synonymous (see Chapter 15). The microdeletion is detectable by fluorescent in-situ hybridization (FISH) technique.6,12–14 Presence of a right aortic arch in TA is associated with higher probability of positive 22q11 microdeletion.15 DiGeorge syndrome patients have additional dysmorphic features such as characteristic facies, thymic and parathyroid hypoplasia, skeletal and renal anomalies, and developmental delay; immunodeficiency and neonatal hypocalcemia are also seen. Most cases occur de novo and only 6% to 10% of the cases are inherited from parents.16 If one of the parents carries the gene deletion, there is 50% chance of recurrence in the next child.
Clarification of Terms Used in the Literature
Aortopulmonary Window versus TA
Developmentally, aortopulmonary (AP) window is also a condition resulting from a defect in the formation of the spiral septum that separates the 2 great arteries. However, in AP window, the defect is in the fusion of the cushions of spiral septum. Improper fusion of spiral septal cushions lead to a defect or communication between ascending aorta (AAo) and MPA. In AP window, both aortic valve and pulmonary valve (PV) are separate and present whereas, TA has only one semilunar valve representing both aortic and PVs.
Pulmonary atresia with VSD
This is also described as TOF with pulmonary atresia. Confusion between TA and PA with VSD occurs due to the terms such as “pseudotruncus” and “hemitruncus” that have been used in the past and were considered as types of TA. These lesions are now considered part of the PA with VSD and not TA as described below. Use of these terms is discouraged.4,17
Pseudotruncus
This term refers to type IV truncus in the Collett and Edwards classification in which PAs originate from descending thoracic Ao. This also refers to the subtype of pulmonary atresia with VSD where PAs are discontinuous and arise from aortic arch and/or descending thoracic Ao. These may be AP collateral arteries rather than native PAs.
Hemitruncus
When one PA arises from AAo and the other PA is either absent or replaced by an AP collateral artery, it was previously called hemitruncus. This also probably is a subtype of PA with VSD with major AP collateral arteries rather than a subtype of TA.
Natural History
Natural history for survival without intervention is poor in babies with TA. Without surgical repair, only 50% survive beyond 1 month, 30% survive beyond 3 months, 18% survive beyond 6 months and 12% survive beyond 1 year.7 Cause of death during the neonatal period is congestive heart failure from large left-to-right shunting and/or truncal valve regurgitation. Some patients develop endocarditis or brain abscess, causing their death.10 Children who survive to one year of age do so because of PA stenosis and, therefore, partial or complete protection of the pulmonary vasculature from exposure to systemic arterial pressure and consequent decreased risk for development of pulmonary hypertensive vascular changes. Therefore, there is low mortality after one year in this subgroup of patients. Very few patients survive infancy and early childhood with significant left-to-right shunting and yet, not develop pulmonary vascular disease. This constitutes half of survivors beyond one year or <5% of all infants born with TA.18 When pulmonary vascular obstructive disease (pulmonary vascular resistance (PVR) > 8 Woods units m2) develops during infancy or later, there is good chance for such patients to survive into their teens without surgical repair.19 However, pulmonary vascular obstructive disease progresses gradually and Eisenmenger syndrome eventually occurs, leading to death.
Clinical Features
History
Clinical presentation in newborn depends upon presence or absence of associated lesions. Commonly, TA babies without any associated lesions present initially some cyanosis due to high PVR at birth. Slowly, cyanosis resolves over several days with development of increase in pulmonary blood flow (PBF). Signs of increased PBF such as tachypnea, chest retractions, irritability and increased sweating will develop. Feeding difficulty and failure to gain weight will ensue. When there is significant stenosis of branch PAs, these symptoms will not occur. Instead, an asymptomatic murmur will be present.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


