Epidemiologic and clinical studies suggest that elevated triglyceride levels are a biomarker of cardiovascular (CV) risk. Consistent with these findings, recent genetic evidence from mutational analyses, genome-wide association studies, and Mendelian randomization studies provide robust evidence that triglycerides and triglyceride-rich lipoproteins are in the causal pathway for atherosclerotic CV disease, indicating that they may play a pathogenic role, much like low-density lipoprotein cholesterol (LDL-C). Although statins are the cornerstone of dyslipidemia management, high triglyceride levels may persist in some patients despite statin therapy. Several triglyceride-lowering agents are available, including fibrates, niacin, and omega-3 fatty acids, of which prescription omega-3 fatty acids have the best tolerability and safety profile. In clinical studies, omega-3 fatty acids have been shown to reduce triglyceride levels, but products containing both eicosapentaenoic acid and docosahexaenoic acid may increase LDL-C levels. Icosapent ethyl, a high-purity eicosapentaenoic acid–only product, does not raise LDL-C levels and also reduces triglyceride, non–high-density lipoprotein cholesterol, and triglyceride-rich lipoprotein levels. In conclusion, omega-3 fatty acids are currently being evaluated in large CV outcome studies in statin-treated patients; these studies should help to elucidate the causative role of triglycerides in atherosclerotic CV disease.
Epidemiologic studies have demonstrated that elevated triglyceride levels are independently associated with increased cardiovascular (CV) risk. Consistent with the epidemiologic data, clinical studies have shown that reaching target triglyceride levels correlates with reduced CV risk. Moreover, triglyceride lowering has been associated with reduced CV risk in certain patients with high baseline triglyceride levels. Together, these data infer that elevated triglyceride levels are a biomarker of CV risk. However, accumulating evidence reviewed herein suggests that triglycerides and triglyceride-rich lipoproteins (TRLs) are in the causal pathway of atherosclerotic CV disease (ASCVD), indicating that they play a pathogenic role in atherosclerosis rather than simply serving as a biomarker of disease risk. This study provides an overview of TRL metabolism, describes genetic evidence supporting the role of triglycerides and TRLs in the causal pathway of ASCVD, and discusses the relevance of these data for managing patients with dyslipidemia.
Overview of TRL Metabolism
Triglycerides are major components of TRLs, including very-low-density lipoprotein (VLDL) and chylomicrons, which are synthesized and secreted from the liver and intestinal enterocytes, respectively ( Figure 1 ). Lipoprotein lipase (LPL) promotes hydrolysis of the core triglycerides in VLDL and chylomicrons, producing VLDL remnants and chylomicron remnants, both of which are enriched in cholesterol relative to triglycerides. Some of these particles may be taken up by the liver and cleared; those that remain in circulation are further modified by LPL and hepatic lipase, resulting in formation of cholesterol-enriched LDL particles. In the clinical setting, non–high-density lipoprotein cholesterol (non–HDL-C) reflects the cholesterol content found in all potentially atherogenic lipoproteins but principally in LDL and in the VLDL remnants and chylomicron remnants. Therefore, to calculate remnant cholesterol (the cholesterol content found in the VLDL remnants and chylomicron remnants), the LDL cholesterol (LDL-C) level may be simply subtracted from the non–HDL-C level. As with LDL particles, VLDL remnants and chylomicron remnants can be taken up by macrophages in the arterial wall, where they may contribute to vascular inflammation and atherosclerotic plaque development and progression; however, unlike LDL particles, remnant particles do not require oxidative modification for this to occur.
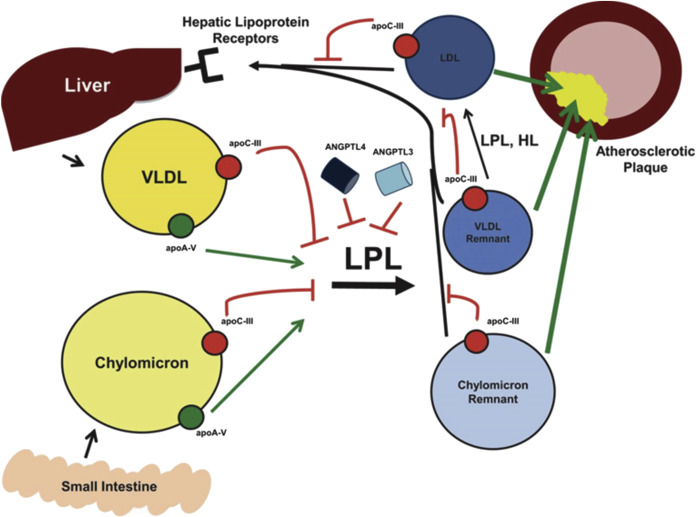
Several apolipoproteins (Apos) are components of VLDL and chylomicrons and are important in TRL metabolism. ApoC-II is a cofactor necessary for full activation of LPL, whereas ApoA-V appears to enhance LPL activity, possibly by facilitating interactions between LPL and TRLs. In contrast, ApoC-III inhibits LPL and may also contribute to hepatic VLDL assembly and secretion. The angiopoietin-like proteins ANGPTL3 and ANGPTL4 are secreted proteins that also inhibit LPL activity, potentially leading to increased triglyceride levels. Importantly, genetic alterations in these factors may have a substantial impact on TRL metabolism and triglyceride levels, and in turn, on atherosclerotic processes and CV outcomes. In addition, recent data from the Jackson Heart Study demonstrated that the ApoA-I/remnant ratio was strongly associated with coronary heart disease incidence.
Genetic Evidence for Triglycerides and TRLs in the Causal Pathway of ASCVD
Genetic variants in LPL and Apos involved in TRL metabolism have been identified and allow further evaluation of whether triglycerides and TRLs are involved in the causal pathway of ASCVD ( Tables 1 to 3 ). Genetic data may be considered particularly compelling for evaluating whether a lipid biomarker is causally related to disease. Evaluation as to whether specific individual genetic loci are causally related to disease can overcome limitations associated with establishing causal inferences based on observational epidemiologic studies (e.g., reverse causation arising from interrelations among LDL-C, HDL-C, and triglycerides). Moreover, genetic data are not limited by confounding design issues that may complicate clinical studies, such as the use of inappropriate study populations, insufficient magnitude of triglyceride lowering, and limited statistical power.
| Reference | N | Variable | Effect on TGs | Effect on Cardiovascular Outcome |
|---|---|---|---|---|
| Pollin 2008 | 809 | ApoC-III R19X null mutation | –46% ( P =4 × 10 -13 ) | CAC: OR=0.35 (95% CI: 0.21–0.60; P =0.002) |
| Jorgensen 2014 | 75,725 | ApoC-III null mutations | –44% ( P <0.001) | IVD: HR=0.59 (95% CI: 0.41–0.86; P =0.007) IHD: HR=0.64 (95% CI: 0.41–0.99; P =0.04) |
| Crosby 2014 | 3,734 | ApoC-III null mutations | –39% ( P =6 × 10 -9 ) | CHD: OR=0.60 (95% CI: 0.47–0.75; P =4 × 10 -6 ) |
| Natarajan 2015 | 6,699 | ApoC-III null mutations | –43.7% ( P =1.83 × 10 -21 ) | CAC: –27.9 U (95% CI: –51.1 to –4.7; P =0.019) |
| Do 2015 | 13,432 | ApoA-V mutations | +61% ( P =0.007) | MI/CAD: OR=2.2 ( P =5 × 10 -7 ) |
| Sarwar 2010 | 302,430 | ApoA-V variant | +16% ( P =4.4 × 10 -24 ) | CHD: HR=1.10 (95% CI: 1.08–1.12; P =2.6 × 10 -7 ) |
| Dewey 2016 | 42,930 | ANGPTL4 variants | –13% ( P =2×10 -23 ) | CAD: OR=0.81 (95% CI: 0.70–0.92; P =0.002) |
| Stitziel 2016 | 120,575 | ANGPTL4 variants | –0.3 standard deviations per allele | CAD: OR=0.86 ( P =4.0 × 10 -8 ) ∗ |
| Reference | N | Study Type | Key Results |
|---|---|---|---|
| Teslovich, 2010 | 96,598 ∗ | Meta-analysis of 46 lipid GWAS | Identified 95 loci, including regions encoding ANGPTL3 and ANGPTL4; 4 novel CAD-associated loci related to HDL-C or TGs but not LDL-C |
| Schunkert 2011 | 147,733 | Meta-analysis of 14 GWAS of CAD | Identified 13 novel loci associated with 6 to 17% increase in risk of CAD; included regions associated with increased TGs |
| Willer 2013 | 188,578 | Linear regression of 149 lipid SNPs | TG effect size correlated with CAD (Pearson r=0.46; P =0.02) |
| Do 2013 | 188,577 | Multivariate analysis of 185 lipid SNPs | TG effect size correlated with CHD after adjusting for LDL-C and HDL-C effect sizes ( P =1 x 10 -9 ) |
| Reference | N | Variable | Effect on TGs | Effect on Cardiovascular Outcome |
|---|---|---|---|---|
| Jorgensen 2013 | 60,113 | 10 combinations of 3 APOA5 variants | Stepwise increase up to 56% | MI: causal OR=1.94 (95% CI: 1.40–1.85); observational OR=1.57 (95% CI: 1.32–2.68) ∗ |
| Varbo 2013 | 73,513 | 3 non-fasting cholesterol-increasing alleles | Correlated with remnant cholesterol (r 2 =0.96) | IHD: causal OR=2.8 (95% CI: 1.9–4.2); observational OR=1.4 (95% CI: 1.3–1.5) † |
| Varbo 2013 | 60,608 | 6 non-fasting cholesterol-increasing alleles | Not measured | IHD: causal OR=3.3 (95% CI: 2.1–5.2); observational OR=1.3 (95% CI: 1.2–1.4) † |
| Thomsen 2014 | 10,208 | 4 LPL genetic variants | Stepwise decrease up to 31% ‡ | All-cause mortality: HR=0.86 for 6 LPL alleles vs 0–3 LPL alleles (95% CI: 0.83–0.88) |
| Holmes 2015 | 62,199 ¶ | 67 triglyceride-related SNPs | Mean increase of 28.5% | CHD § : OR (unrestricted)=1.62 (95% CI: 1.24– 2.11); OR (restricted)=1.61 (95% CI: 1.00–2.59) |
∗ For doubling of nonfasting triglyceride levels.
† For 1 mmol/L (39 mg/dl) increase in remnant cholesterol levels.
‡ With increasing number of LPL alleles.
§ Unrestricted allele scores were based on all 67 triglyceride-related SNPs; restricted allele scores were for 1-log increment in triglyceride level and included only 27 triglyceride-related SNPs that were not also associated with LDL-C or HDL-C.
Mutational analyses/exome sequencing
Early studies found that mutations conferring reduced TRL metabolism are associated with atherosclerosis and CHD, whereas those enhancing TRL metabolism have the opposite clinical effect. Several substitution mutations have been identified at the N-terminal end of LPL that reduce its catalytic activity. In a meta-analysis, carriers of the Gly188Glu substitution mutation in LPL had reduced LPL activity, an average increase in triglyceride levels of 78%, and a nearly fivefold increased CHD risk compared with noncarriers. The effects of the Asp91Asn and Asn291Ser mutations on LPL activity and triglyceride levels were smaller, and they had only a borderline effect on CHD risk. Loss-of-function mutations in ApoC-II, the activating cofactor for LPL, have been reported to cause profound increases in serum triglyceride levels leading to chylomicronemia and pancreatitis. Similarly, mutations in the gene encoding ApoA-V have been associated with increased triglyceride levels as well as higher risk of myocardial infarction (MI)/coronary artery disease and increased risk of CHD ( Table 1 ).
Mutations in APOC3 have the opposite effect, consistent with the inhibitory effect of this Apo on LPL activity. In a cohort of Amish subjects, carriers of an R19X mutation in APOC3 had a 46% reduction in serum triglyceride levels compared with noncarriers, which was associated with a 65% reduction in risk of coronary artery calcification, a measure of subclinical atherosclerosis (p = 0.002). Consistent with these findings, rare loss-of-function APOC3 mutations found through exome sequencing were associated with decreased triglyceride levels and CHD risk compared with noncarriers in 2 recent studies ( Table 1 ). Similarly, a recent analysis of APOC3 mutations in subjects of European, African, Asian, and Hispanic ancestries also found that carriers had reduced plasma triglycerides and decreased burden of coronary artery calcification ( Table 1 ).
Most recently, ANGPTL4 mutations were shown to be associated with lower triglyceride levels and lower risk of coronary artery disease ( Table 1 ).
Genome-wide association studies
Genome-wide association studies are designed to search large cohorts for common genetic variants, such as single nucleotide polymorphisms (SNPs) that occur more frequently in patients with a certain disease compared with those without the disease, to identify susceptibility genes that may contribute to that disease. Genome-wide association studies have been conducted to identify susceptibility loci for CHD as well as specific lipid traits that have been associated with CHD; these studies detected susceptibility loci in gene regions encoding ApoC-III, ApoA-V, ANGPTL3, and ANGPTL4 ( Table 2 ). In the genome-wide association studies evaluating lipid traits, a linear regression analysis was performed to evaluate how lipid levels were related to CHD risk. Notably, significant associations with CHD risk were found for LDL-C effect size (Pearson r = 0.74; p = 7 × 10 −6 ) and triglyceride effect size (Pearson r = 0.46; p = 0.02) but not for HDL-C effect size ( r = 0.0009; p = 0.99). Because most SNPs affect multiple lipid fractions, a multivariate analysis was performed on the 185 SNPs that had moderate to strong effects on triglyceride levels; the triglyceride effect size was strongly associated with CHD after adjusting for both LDL-C and HDL-C effect sizes ( Table 2 ). Similar analyses showed that LDL-C (p = 2 × 10 −22 ), but not HDL-C (p = 0.35), was associated with CHD after correcting for the other corresponding lipids.
Extending our understanding of the genetic contributions of lipid-related traits beyond the genomic studies of DNA sequence variants, an epigenome-wide association study examined the relation between fasting blood lipid levels and differential methylation sites. In this study, methylation at 4 sites of intron 1 of carnitine palmitoyltransferase 1A was found to be strongly associated with triglyceride and VLDL cholesterol levels and may represent novel loci involved in the prevention of CVD.
Mendelian randomization studies
Mendelian randomization studies are based on the segregation and independent assortment of specific genotypes according to the laws of Mendelian genetics. In these studies, subjects with specific genotypes that predispose to a specific effect (e.g., hypertriglyceridemia) are compared with subjects without those genotypes. Because these studies are not affected by confounding factors seen in observational epidemiologic studies, they can provide robust evidence for the causal involvement of a specific mechanism in a disease state. Mendelian randomization studies have been conducted that evaluate whether factors involved in TRL metabolism are causally associated with atherosclerosis and CHD ( Table 3 ).
Several Mendelian randomization studies were conducted using data from 3 Copenhagen population cohorts. In the first study, 3 common APOA5 variants were used to define 10 common genotype combinations, which were associated with substantial stepwise increases in nonfasting triglyceride levels and calculated remnant cholesterol levels (which is the cholesterol content of TRLs), and with corresponding increases in risk of MI. For a doubling of nonfasting triglyceride levels and calculated remnant cholesterol levels, the causal odds ratios for risk of MI were 1.94 (95% CI 1.40 to 1.85) and 2.23 (95% CI 1.48 to 3.35), respectively. Studies 2 and 3 evaluated genotypes associated with increasing nonfasting remnant cholesterol levels and differed based on which APOA5 and LPL genotypes were used to define increasing remnant cholesterol levels. Both studies found that remnant cholesterol levels were highly correlated with nonfasting triglyceride levels and with observed CHD risk ( Table 3 ). A subsequent Mendelian randomization study using data from the Copenhagen City Heart Study found that genetic variants in LPL resulted in reduced triglyceride levels, and that a higher number of triglyceride-decreasing LPL alleles was associated with increased survival (p = 0.004).
To overcome limitations in which only a limited number of variants are used, a Mendelian randomization meta-analysis was conducted using data from 17 studies involving 62,199 participants and 12,099 CHD events. Unrestricted allele scores were determined based on all SNPs that had established associations with triglycerides, HDL-C, or LDL-C, and then restricted allele scores were generated in which SNPs were excluded if they were also associated with either of the 2 other lipids. For triglycerides, both the unrestricted allele score based on 67 SNPs and the restricted allele score based on 27 SNPs were significantly associated with CHD ( Table 3 ). Allele scores for LDL-C but not HDL-C were significantly associated with CHD. However, for HDL-C, the restricted allele score was not associated with CHD; the odds ratio for a 1 mmol/L (39 mg/dl) genetic increment in HDL-C level was 0.91 (95% CI 0.42 to 1.98).
Taken together, the genetic data for triglycerides and TRLs align with the epidemiologic and clinical data and support a causative role for these lipids in ASCVD and compare favorably with those linking LDL-C with ASCVD, in contrast to those for HDL-C that did not show a causative role in CV disease. The latter finding is notable given that HDL-C–increasing drugs have failed to improve CV outcomes in large clinical studies.
Clinical implications
Statins are widely used as an adjunct to diet and exercise in the first-line treatment of patients with lipid abnormalities. Statins substantially reduce LDL-C levels and may also help to lower triglyceride levels. However, the effects of statins on triglyceride levels are often insufficient, and therefore a triglyceride-lowering agent may need to be added to statin therapy if triglyceride levels remain high. Treatment options include omega-3 fatty acids, niacin, and fibrates.
Several prescription omega-3 fatty acid products are available; each is currently approved as an adjunct to diet for reducing triglyceride levels in adult patients with severe hypertriglyceridemia (≥500 mg/dl). Most prescription omega-3 fatty acid products (omega-3-acid ethyl esters [Lovaza; GlaxoSmithKline, Research Triangle Park, North Carolina]; omega-3-acid ethyl esters A [Omtryg; Trygg Pharma, Arlington, Virginia]; and omega-3-carboxylic acids [Epanova; AstraZeneca, Wilmington, Delaware]) contain a mixture of eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). However, icosapent ethyl (Vascepa; Amarin Pharma Inc., Bedminster, New Jersey) is a high-purity prescription formulation containing the ethyl ester of EPA. In clinical trials, all prescription omega-3 fatty acids significantly reduced triglyceride levels in hypertriglyceridemic patients in the absence or presence of background statin therapy. Notably, the products containing both EPA and DHA were found in clinical trials to increase LDL-C levels, an unwanted potential effect in treating patients with lipid abnormalities. In contrast, icosapent ethyl did not increase LDL-C levels compared with placebo in clinical trials. Icosapent ethyl, as well as products containing both EPA and DHA, has also shown beneficial effects on the levels of other atherogenic lipid parameters discussed herein, including non–HDL-C, VLDL cholesterol, and/or TRLs.
Omega-3 fatty acid prescription products have well-established favorable safety and tolerability profiles. In contrast, niacin and fibrates, although effective in reducing triglyceride levels and other lipid parameters, are associated with tolerability issues that may compromise treatment. Niacin causes flushing, which many patients find intolerable, and also has the potential to increase serum glucose and cause liver toxicity or myopathy, particularly when coadministered with a statin. Fibrates may blunt the beneficial effects of statins on LDL-C, particularly in patients with atherogenic dyslipidemia, and like niacin, has the potential to cause myopathy when used in combination with a statin. The use of niacin or fibrates in combination with a statin has recently been removed from the labels of several products because of the lack of data showing CV benefit with these combinations.
The benefits of omega-3 fatty acids may extend beyond triglyceride lowering, including effects on vascular and cardiac hemodynamics, arrhythmia, inflammation, endothelial function, thrombosis, and production of inflammation-resolving mediators such as resolvins and protectins. Although both EPA and DHA may contribute to health benefits of omega-3 fatty acids, it is also important to note that EPA, as a 20-carbon fatty acid, but not DHA (a 22-carbon fatty acid), can displace arachidonic acid (also a 20-carbon fatty acid) in metabolic pathways and thus antagonize arachidonic-mediated inflammatory reactions. Indeed, EPA specifically has been shown to have pleiotropic effects that beneficially influence multiple steps in atherosclerosis development and progression including endothelial dysfunction, oxidative stress, foam cell formation, inflammation and cytokine effects, plaque formation and progression, platelet aggregation, thrombus formation, and plaque rupture.
Recent studies support the concept that add-on therapy to a statin can reduce residual risk and provide CV benefit. Two large outcome trials are testing the triglyceride-lowering hypothesis with prescription omega-3 fatty acid products in statin-treated patients at high CV risk. The Reduction of Cardiovascular Events with EPA-Intervention Trial (REDUCE-IT; NCT01492361 ) is evaluating 4 g/day icosapent ethyl (prescription EPA only), whereas the Statin Residual Risk Reduction With Epanova in High CV Risk Patients With Hypertriglyceridemia study (STRENGTH; NCT02104817 ) is evaluating omega-3-carboxylic acids (prescription EPA plus DHA). The primary end point is a composite including CV death, MI, stroke, revascularization, and hospitalization for unstable angina. Data from these clinical outcome trials will help to elucidate whether high-dose prescription omega-3 fatty acid products reduce ASCVD in high-risk, statin-treated patients, such as those commonly encountered in clinical practice.
Acknowledgment
Medical review, scientific reference checks, and associated assistance were provided by Joy Bronson and Christina Copland, PhD, MPH, of Amarin Pharma Inc., Bedminster, New Jersey, USA. Medical writing assistance was provided by Peloton Advantage, LLC, Parsippany, New Jersey, USA, and was funded by Amarin Pharma Inc .
Disclosures
Dr. Budoff has received research/grant support from General Electric and is a member of the speaker’s bureau for Amarin Pharma Inc.
See page 142 for disclosure information.
Genetic Evidence for Triglycerides and TRLs in the Causal Pathway of ASCVD
Genetic variants in LPL and Apos involved in TRL metabolism have been identified and allow further evaluation of whether triglycerides and TRLs are involved in the causal pathway of ASCVD ( Tables 1 to 3 ). Genetic data may be considered particularly compelling for evaluating whether a lipid biomarker is causally related to disease. Evaluation as to whether specific individual genetic loci are causally related to disease can overcome limitations associated with establishing causal inferences based on observational epidemiologic studies (e.g., reverse causation arising from interrelations among LDL-C, HDL-C, and triglycerides). Moreover, genetic data are not limited by confounding design issues that may complicate clinical studies, such as the use of inappropriate study populations, insufficient magnitude of triglyceride lowering, and limited statistical power.



