Chapter 26 Transplantation Immunobiology and Immunosuppression
Transplantation has revolutionized the treatment of end-stage organ failure. Today, there are over 25,000 transplantations performed annually and more than 100,000 patients are currently listed and awaiting an organ. The concept of tissue transplantation is certainly not new. As early as 800 BC, skin grafts were performed in India to conceal amputation of the nose, a punishment for adultery. History also is replete with legends and myths recounting the replacement of limbs and organs. One of the first references specifically to solid organ replacement as a therapeutic solution occurred when Tua-Ho, of China, reportedly replaced diseased organs with healthy ones approximately 200 AD. A more well-known myth of early transplantation derived from the miracle of Saints Cosmas and Damian (brothers and subsequently patron saints of physicians and surgeons), in which they successfully replaced the gangrenous leg of the Roman deacon Justinian with a leg from a recently deceased Ethiopian (Fig. 26-1). However, it was not until the French surgeon Alexis Carrel developed a method for joining blood vessels in the late 19th century that the transplantation of organs became technically feasible and verifiable accounts of transplantation began (Fig. 26-2). He was awarded the Nobel Prize in Medicine in 1912 “in recognition of his work on vascular suture and the transplantation of blood vessels and organs.” Having established the technical component, Carrel himself noted that there were two issues to be resolved “regarding the transplantation of tissues and organs . . . the surgical and the biological.” He had solved one aspect, the surgical, but he also understood that “it will only be through a more fundamental study of the biological relationships existing between living tissues”1 that the more difficult problem of the biology would come to be solved. Forty years would pass before another set of eventual Nobel Prize winners, including Peter Medawar, would begin to define the process whereby one individual rejects another’s tissue (Fig. 26-3).2 Shortly thereafter, Joseph Murray, Nobel Laureate 1990, performed the first successful renal transplant between identical twins in 1954 (Fig. 26-4).3 At the same time, Gertrude Elion, who worked as an assistant to George Hitchings at Wellcome Research Laboratories, developed several new immunosuppressive compounds, including 6-MP and Aza. Murray and Roy Calne subsequently introduced these agents into clinical practice, permitting nonidentical transplantation to be successful. Elion and Hitchings later shared the Nobel Prize in 1988 for their work on “the important principles of drug development.” The subsequent discovery of increasingly potent agents to suppress the rejection response has led to the success in allograft survival that we enjoy today. It is this collaboration between scientists and surgeons that has driven our understanding of the immune system as it relates to transplantation. In this chapter, we will provide a primer on rejection in the context of the broader immune response, review the specific agents that are used to suppress the rejection response, and provide a glimpse into the future of the field of immunology and the immune response.

FIGURE 26-2 Triangulation technique of vascular anastomosis by Alexis Carrel.
(From Edwards WS, Edwards PD: Alexis Carrel: Visionary surgeon, Springfield, Ill, 1974, Charles C Thomas, pp 64–83.)

FIGURE 26-3 Sir Peter Medawar.
(Courtesy Bern Schwartz Collection, National Portrait Gallery, London.)
The Immune Response
The immune system has evolved to include two complementary divisions to respond to disease, the innate and acquired immune systems. Broadly speaking, the innate immune system recognizes general characteristics that have through selective pressure come to represent universal pathologic challenges to our species (e.g., ischemia, necrosis, trauma, certain nonhuman cell surfaces).4 Conversely, the acquired arm recognizes specific structural aspects of foreign substances, usually peptide or carbohydrate moieties, recognized by receptors generated randomly and selected to avoid self-recognition. Although the two systems differ in their specific responsibilities, they act in concert to influence each other to achieve an optimal overall response.
Acquired Immunity
Human Histocompatibility Complex
The antigens primarily responsible for human allograft rejection are those encoded by the HLA region of chromosome 6 (Fig. 26-5). The polymorphic proteins encoded by this locus include class I molecules (HLA-A, -B, and -C) and class II molecules (HLA-DR, -DP, and -DQ). There are additional class I genes with limited polymorphism (E, F, G, H, and J) but they are not currently used in tissue typing for transplantation and are not considered here. There are class III genes as well, but they are not cell surface proteins involved in antigen presentation directly but include molecules pertinent to the immune response by various mechanisms—tumor necrosis factors-α (TNF-α) and TNF-β, components of the complement cascade, nuclear transcription factor β, and HSP 70. Other conserved genes in HLA include genes necessary for class I and class II presentation of peptides, such as the peptide transporter proteins TAP-1 and TAP-2 and proteosome proteases LMP-2 and LMP-7.5 Although other polymorphic genes, referred to as minor histocompatibility antigens, exist in the genome outside of the HLA locus, they play a more limited role in transplant rejection and will not be covered here. It is, however, important to point out that even HLA-identical individuals are subject to rejection on the basis of these minor differences. The blood group antigens of the ABO system must also be considered transplantation antigens and their biology is critical to humoral rejection.
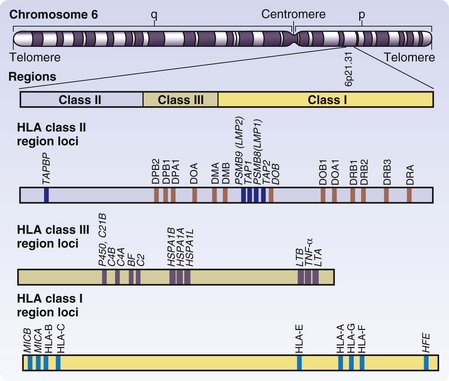
Each class I molecule is encoded by a single polymorphic gene that is combined with the nonpolymorphic protein β2-microglobulin (β2M; chromosome 15) for expression. The polymorphism of each class I molecule is extreme, with 30 to 50 alleles/locus. Class II molecules are made up of two chains, α and β, and individuals differ not only in the alleles represented at each locus, but also in the number of loci present in the HLA class II region. The polymorphism of class II is thus increased by combinations of α and β chains, as well as of hybrid assembly of chains from one class II locus to another. As the HLA sequence varies, the ability of various peptides to bind to the molecule and be presented for T cell recognition changes. Teleologically, this extreme diversity is thought to improve the likelihood that a given pathogenic peptide will fit into the binding site of these antigen-presenting molecules, thus preventing a single viral agent from evading detection by T cells of an entire population.6
Class I Major Histocompatibility Complex
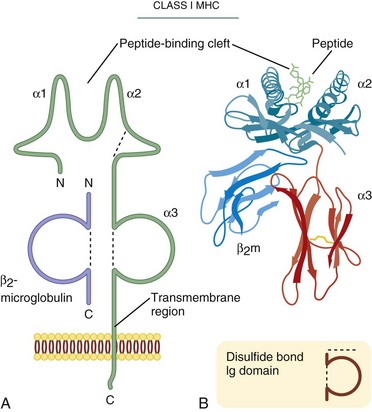
The three-dimensional structure of class I molecules (HLA-A, -B, and -C) was first elucidated in 1987.7 The class I molecule is composed of a 44-kDa transmembrane glycoprotein (α chain) in a noncovalent complex with a nonpolymorphic 12-kDa polypeptide called β2-M. The α chain has three domains, α-1, α-2, and α-3. The critical structural feature of class I molecules is the presence of a groove formed by two α helices mounted on a β-pleated sheet in the α-1 and α-2 domains (Fig. 26-6). Within this groove, a nine-amino-acid peptide, formed from fragments of proteins being synthesized in the cell’s endoplasmic reticulum, is mounted for presentation to T cells. Almost all the significant sequence polymorphism of class I is located in the region of the peptide-binding groove and in areas of direct T cell contact. The assembly of class I is dependent on association of the α chain with β2-M and native peptide within the groove. Incomplete molecules are not expressed. In general, all peptides made by a cell are candidates for presentation, although sequence alterations in this region favor certain sequences over others. The α-3 immunoglobulin-like domain, which is the domain closest to the membrane and interacts with the CD8 molecule on the T cell, demonstrates limited polymorphism and is conserved to preserve interactions with CD8+ T cells.
Human class I presentation occurs on all nucleated cells and expression can be increased by certain cytokines, thus allowing the immune system to inspect and approve of ongoing protein synthesis. Interferons (IFN-α, IFN-β, and IFN-γ) induce an increase in the expression of class I molecules on a given cell by increasing levels of gene expression. T cell activation occurs when a given T cell encounters a class I MHC molecule carrying a peptide from a nonself protein presented in the proper context (e.g., viral protein is processed in an infected cell and the peptide fragments are presented on class I molecules for T cell recognition). So-called cross-presentation may also occur, in which certain antigen-presenting cells (APCs)—namely, a subset of dendritic cells— have the ability to take up and process exogenous antigen and present it on class I molecules to CD8+ T cells.8 In the case of transplantation, this activation is not only possible when foreign peptide is identified after the donor MHC has been processed and presented on recipient APCs, but more commonly occurs when a T cell interacts directly with the nonself class I MHC, the so-called direct alloresponse.
Class II MHC
The three-dimensional structure of class II molecules was inferred by sequence homology to class I in 1988 and eventually proven by x-ray crystallography in 1993 (Fig. 26-7).9 The class II molecules contain two polymorphic chains, one approximately 32 kDa and the other approximately 30 kDa. The peptide-binding region is composed of the α-1 and β-1 domains. The immunoglobulin-like domain is composed of the α-2 and β-2 segments. Similar to the class I immunoglobulin-like α-3 domain, there is limited polymorphism in these segments and the β-2 domain, in particular, is involved in the binding of the CD4 molecule, helping to restrict class II interactions to CD4+ T cells. Class II molecule assembly requires association of both the α chain and β chains in combination with a temporary protein called the invariant chain.10 This third protein covers the peptide-binding groove until the class II molecule is out of the endoplasmic reticulum and is sequestered in an endosome. Proteins that are engulfed by a phagocytic cell are degraded at the same time as the invariant chain is removed, allowing peptides of external sources to be associated with and presented by class II. In this way, the acquired immune system can inspect and approve of proteins that are present in circulation or that have been liberated from foreign cells or pathogens through the phagocytic process. Accordingly, class II molecules, in contrast to class I molecules, are confined to cells related to the immune response, particularly APCs (e.g., macrophages, dendritic cells, B cells, and monocytes). Class II expression can also be induced on other cells, including endothelial cells under the appropriate conditions. After binding class II molecules, CD4+ T cells participate in APC-mediated activation of CD8+ T cells and antibody-producing B cells. In the case of transplanted organs, ischemic injury at the time of transplantation accentuates the potential for T cell activation by the upregulation of class I and class II molecules locally on the recipient. The trauma of surgery and ischemia also upregulate class II molecules on all cells of an allograft, making nonself-MHC more abundant. Host CD4+ T cells may then recognize donor MHC directly (direct alloresponse) or after antigen processing (indirect alloresponse) and then proceed to participate in rejection.

HLA Typing: Implications for Transplantation
For the reasons already discussed, closely matched transplants are less likely to be recognized and rejected than are similar grafts differing by multiple alleles at the MHC. HLA matching has a clear influence on the prolongation of graft survival. Humans have two different HLA-A, -B, and -DR alleles—one from each parent, six in total. Although clearly important, the HLA-C, -DP, and -DQ loci are administratively dismissed in general organ allocation. Although current immunosuppressive regimens negate much of the impact of matching, there have been several studies that have demonstrated improvements in renal allograft survival when the six primary alleles are matched between donor and recipient, a so-called six-antigen match (Fig. 26-8). Historically, MHC match has been defined using two cellular assays, the lymphocytotoxicity assay and the mixed lymphocyte reaction (MLC). Both assays define MHC epitopes but do not comprehensively define the entire antigen or the exact genetic disparity involved. Techniques now exist for precise genotyping via molecular techniques that distinguish the nucleotide sequence of an individual’s MHC.
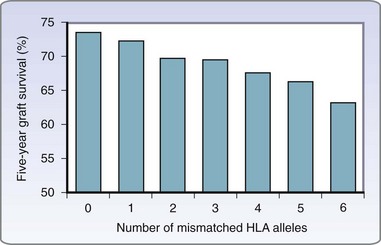
(Data from U.S. Organ Procurement and Transplant Network and the Scientific Registry of Transplant Patients: 2008 OPTN/SRTP annual report: Transplant data 1998–2007 [http://optn. transplant.hrsa.gov/ar2008].)
Cellular Components of the Acquired Immune System
The key cellular components of the immune system, T cells, B cells, and antigen-presenting cells, are hematopoietically derived and arise from a common progenitor stem cell. The development of the lymphoid system begins with pluripotent stem cells in the liver and bone marrow of the fetus. As the fetus matures, the bone marrow becomes the primary site of lymphopoiesis. B cells were named after the primary lymphoid organ that produces B cells in birds, the bursa of Fabricius. In humans and most other mammals, precursor B cells remain within the bone marrow as they mature and fully develop. Although precursor T cells also originate in the bone marrow, they soon migrate to the thymus, the primary site of T cell maturation, where they become “educated” to self and acquire their specific cell surface receptors and the ability to generate effector function. Mature lymphocytes are then released from the primary lymphoid organs, the bone marrow and thymus, to populate the secondary lymphoid organs, including lymph nodes, spleen, and gut, as well as peripheral tissues. Each of these cells has a unique role in establishing the immune response. The highly coordinated network is regulated in part through the use of cytokines (Table 26-1).
Table 26-1 Summary of Cytokines
| CYTOKINE | SOURCE | PRINCIPAL CELLULAR TARGETS AND BIOLOGIC EFFECTS |
|---|---|---|
| IL-1 | Macrophages, endothelial cells, some epithelial cells | |
| IL-2 | T cells | T cells: Proliferation, ↑ cytokine synthesis, survival, potentiate Fas-mediated apoptosis, promote regulatory T cell development |
| IL-3 | T cells | |
| IL-4 | CD4+ T cells (Th2), mast cells | |
| IL-5 | CD4+ T cells (Th2) | |
| IL-6 | Macrophages, endothelial cells, T cells | |
| IL-7 | Fibroblasts, bone marrow stromal cells | |
| TNF | Macrophages, T cells | |
| IFN-γ | T cells (Th1, CD8+ T cells), NK cells | |
| Type I IFNs (IFN-α, IFN-β) | Macrophages, IFN-α; fibroblasts, IFN-β | |
| TGF-β | T cells, macrophages, other cell types | |
| Lymphotoxin (LT) | T cells | |
| IL-8 | Lymphocytes, monocytes | |
| IL-9 | Activated Th2 cells, lymphocytes | |
| IL-10 | Macrophages, T cells (mainly regulatory T cells) | |
| IL-11 | Bone marrow stromal cells | |
| IL-12 | Macrophages, dendritic cells | |
| IL-13 | CD4+ T cells (Th2), NKT cells, mast cells | |
| IL-14 | T cells, some B cell tumors | |
| IL-15 | Macrophages, others | |
| IL-17 | T cells | |
| IL-18 | Macrophages | |
| IL-23 | Macrophages, dendritic cells | |
| IL-27 | Macrophages, dendritic cells |
G-CSF, Granulocyte-colony stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor.
Adapted from Abbas AK, Lichtman AH, Pillai S: Cellular and molecular immunology, ed 6, Philadelphia, 2010, Saunders-Elsevier.
T Cells
T Cell Receptor
Considerable progress has been made in defining the mechanisms of T cell maturation and the development of a functional TCR. The formation of the TCR is fundamental to understanding its function.11 When precursor T cells migrate from the fetal liver and bone marrow to the thymus, they have yet to obtain their specialized TCR or accessory molecules. On arrival to the thymus, T cells undergo a remarkable rearrangement of their DNA, which encodes the various chains of the TCR (α, β, γ, and δ; Fig. 26-9). The order of genetic rearrangement recapitulates the evolution of the TCR. T cells first attempt to recombine the γ and δ TCR genes and then, if recombination fails to yield a properly formed receptor, resort to the more diverse α and β TCR genes. The γδ configuration is typically not successful and thus most T cells are αβ T cells. T cells expressing the γδ TCR have more primitive functions, including recognition of heat shock proteins and activity similar to natural killer (NK) cells, as well as MHC recognition, whereas αβ T cells are more typically limited to recognition of MHC complexed with processed peptide.
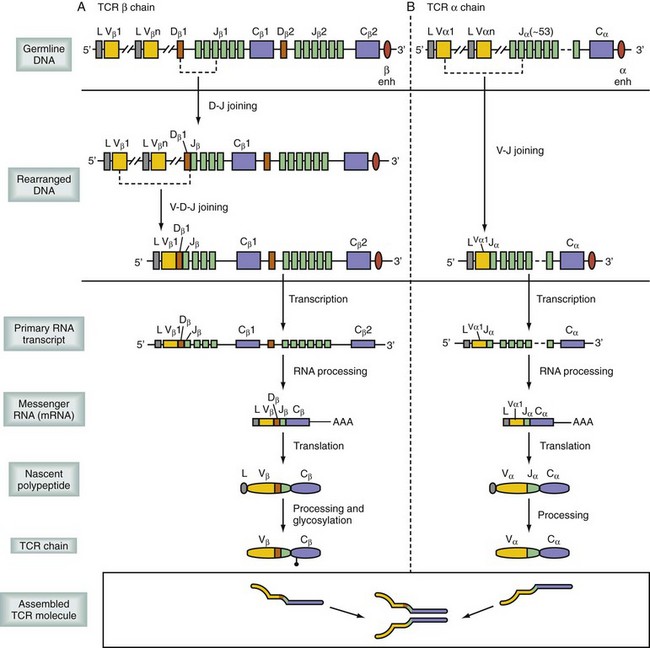
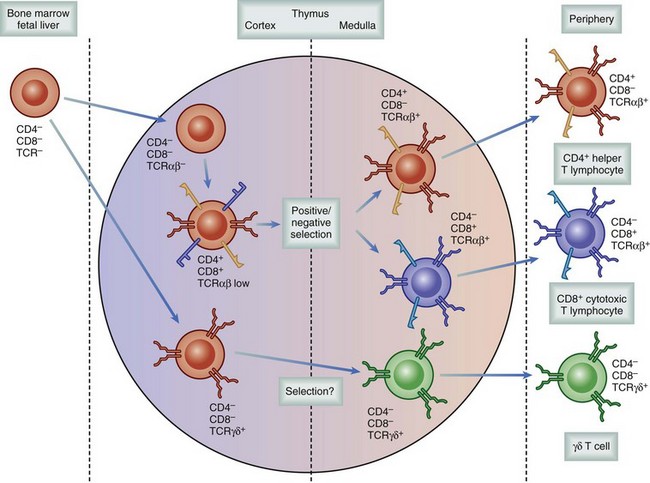
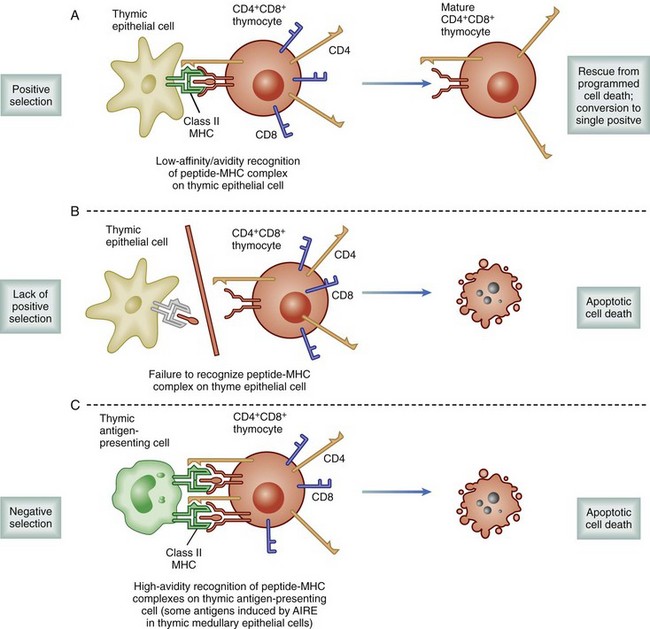
Regardless of the genes used, individual cells recombine to express a TCR with only a single specificity. The rearrangements occur randomly, resulting in a population of T cells capable of binding 109 different specificities, essentially all combinations of MHC and peptide. As a result, the frequency of naïve T cells available to respond to any given pathogen is relatively small, between 1 in 200,000 to 500,000. These developing T cells also express both CD4 and CD8, accessory molecules, which strengthen the TCR binding to MHC. These accessory molecules further increase the binding repertoire of the population to include class I or class II MHC molecules. If the process of T cell maturation ended at this stage, there would be a host of T cells that could recognize self-MHC–peptide complexes, resulting in an uncontrolled global autoimmune response. To avoid the release of autoreactive T cells, developing cells undergo a process following recombination known as thymic selection (Fig. 26-10).12 Cells initially interact with the MHC-expressing cortical thymic epithelium, which produces hormones (thymopoietin and thymosin) as well as cytokines (e.g., IL-7), which are critical to T cell development. If binding does not occur to self-MHC, those cells are useless to the individual—because they cannot bind self cells to assess for viral infection—and they undergo programmed self-destruction via apoptosis, a process called positive selection (Fig. 26-11). Cells surviving positive selection then move to the thymic medulla and, normally, eventually lose CD4 or CD8. If binding to self-MHC in the medulla occurs with an unacceptably high affinity, programmed death again results; this process is called negative selection. The precise nature of this affinity threshold remains a matter of intense investigation and involves interaction with hematopoietic cells that reside in the thymus. The only cells released into the periphery are those that can bind self-MHC and avoid activation. Whereas T cells are restricted to bind self-MHC–peptide complexes without activation, the selection process does not consider foreign MHC. Thus, by random chance, some cells with appropriate affinity for self-MHC survive and have an inappropriately high affinity for the MHC molecules of other individuals. In the setting of transplantation, these recipient T cells are able to recognize donor MHC-peptide complexes because there are sufficient conserved motifs shared between donor and self-MHC molecules. However, because donor MHC was not present during the thymic education process, the binding of donor MHC by an alloreactive T cell leads to activation, and rejection ensues. The precursor frequency or the number of alloreactive T cells is much higher than the 1 in 200,000 or 500,000 T cells available to react toward any given antigen. Because T cells are selected to bind self-MHC, the frequency specific for a similar, nonself-MHC (i.e., alloreactive) is between 1% and 10% of all T cells.
T Cell Activation
T cells can then specifically recognize and destroy cells that make peptide products of mutation or viral infection. Because the number of potential antigens is high, and the likelihood is that self-antigens vary minimally from foreign antigens, the nature of the TCR-binding event has evolved so that a single interaction with an MHC molecule is not sufficient to cause activation. In fact, a T cell must register a signal from approximately 8000 TCR–ligand interactions with the same antigen before a threshold of activation is reached.13 Each event results in the internalization of the TCR. Because resting T cells have low TCR density, sequential binding and internalization over several hours is required. Transient encounters are not sufficient. This threshold is reduced considerably by appropriate costimulation signals (see later).
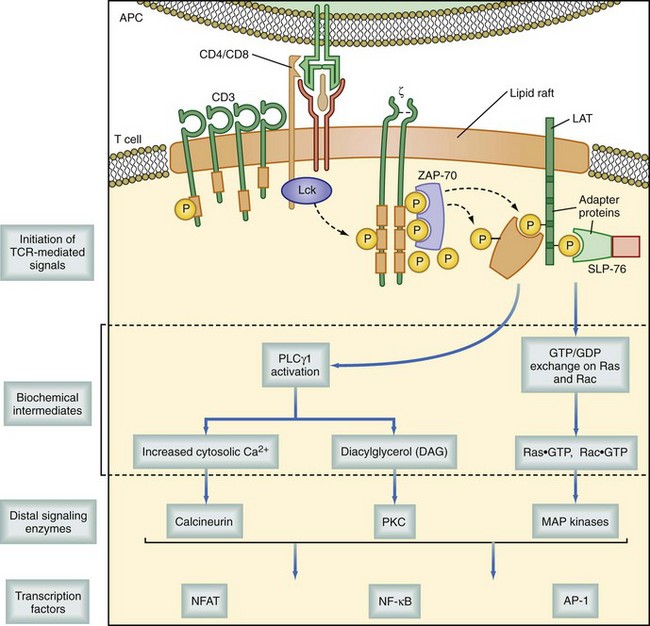
As discussed in the previous section, most TCRs are heterodimers composed of two transmembrane polypeptide chains, α and β. The αβ-TCR is noncovalently associated with several other transmembrane signaling proteins, including CD3 (composed of three separate chains, γ, δ, and ε), and ζ chain molecules, as well as the appropriate accessory molecule from the T cell. This is either CD4 or CD8, which associates with its respective MHC molecule. Together, these proteins are known as the TCR complex. When the TCR is bound to an MHC molecule and the proper configuration of accessory molecules stabilize its binding, a signal is initiated by intracytoplasmic protein tyrosine kinases (PTKs). These PTKs include p56lck (on CD4 or CD8), p59Fyn, and ZAP70; the latter two are associated with CD3. Repetitive binding signals combined with the appropriate costimulation eventually activate phosphokinase C-gamma (PLC-γ1), which in turn hydrolyzes the membrane lipid phosphatidyl inositol biphosphate (PIP2), thereby releasing inositol triphosphate (IP3) and diacyl glycerol (DAG). IP3 binds to the endoplasmic reticulum, causing a release of calcium that induces calmodulin to bind to and activate calcineurin. Calcineurin dephosphorylates the critical cytokine transcription factor nuclear factor of activated T cells (NFAT), prompting it, with the transcription factor nuclear factor κB (NF-κB), to initiate the transcription of cytokines, including IL-2 and its receptor (Fig. 26-12). Resting T cells express only low levels of the IL-2 receptor (IL-2R; CD25) but, with activation, IL-2R expression is increased. As the activated T cell begins to produce IL-2 secondary to events initiated by TCR activation, the cytokine begins to work in autocrine and paracrine fashions, potentiating DAG activation of protein kinase C (PKC). PKC is important in activating many gene regulatory steps critical for cell division. This effect, however, is restricted only to T cells that have undergone activation after encountering their specific antigen, leading to IL-2R expression. Thus, the process limits proliferation and expansion to only those clones specific for the offending antigen. As the antigenic stimulus is removed, IL-2R density decreases and the TCR complex is reexpressed on the cell surface. There is a negative feedback system between the TCR and the IL-2R, resulting in a highly regulated and efficient system that is only reactive in the presence of antigen and ceases to function once antigen is removed. Many of these steps in T cell activation have been targeted in the development of immunosuppressive agents. These will be discussed in detail later in this chapter.
Costimulation
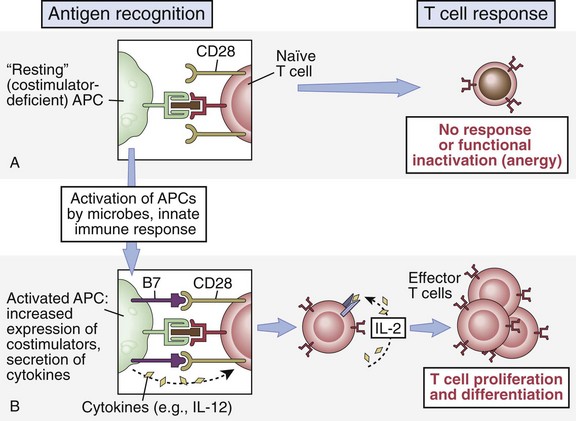
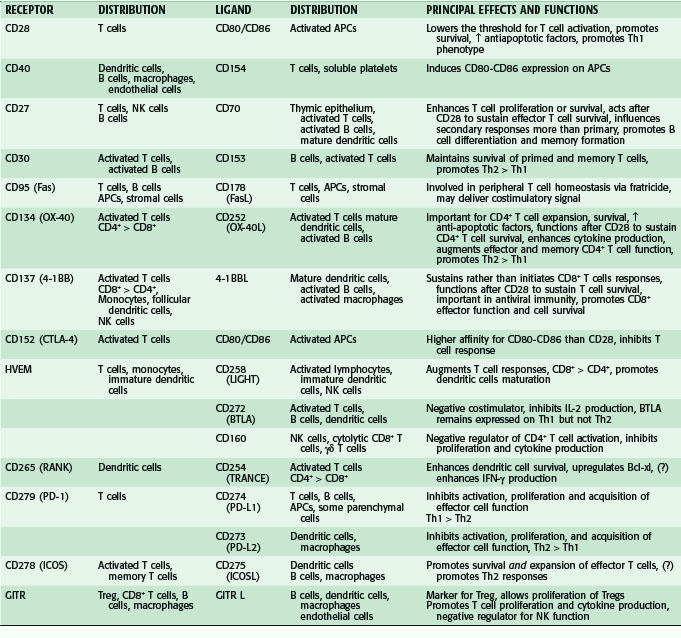
As noted, recognition of the antigenic peptide–MHC complex via TCR binding is usually not sufficient alone to generate a response in a naïve T cell. Additional signals, through so-called costimulatory pathways, are required for optimal T cell activation.14,15 In fact, receipt of TCR complex signaling, often referred to as signal 1, in the absence of costimulation, or signal 2, not only fails to achieve activation but can lead to a state of inaction, or anergy (Fig. 26-13). An anergic T cell is now unable to respond, even if given both appropriate stimuli.16 This characteristic of the immune system is thought to be one of the major mechanisms in tolerance to self-antigens in the periphery, crucial in the prevention of autoimmunity. Researchers have exploited this discovery using antibodies or receptor fusion proteins designed to block interactions between key costimulatory molecules at the time of antigen exposure. Most research to date has focused on the interactions of two costimulatory pathways, the CD28-B7 pathway (immunoglobulin-like superfamily members) and CD40-CD154 pathway (tumor necrosis factor [TNF]–TNFR superfamily members). There have been, however, many additional pairings in these same families and others that have been found to have distinct roles in costimulatory function (Table 26-2).
T Cell Effector Functions
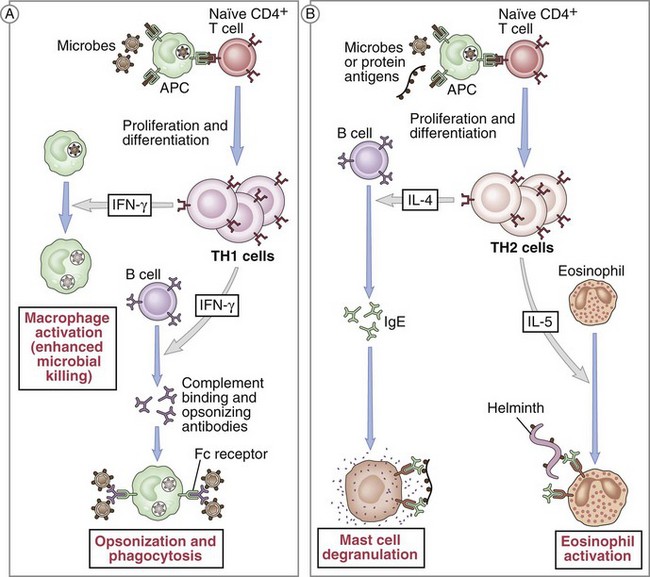
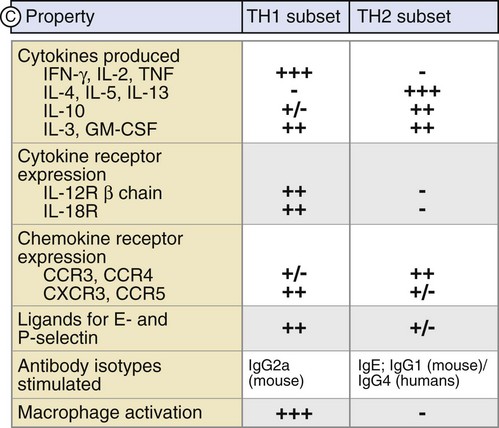
Following activation, CD4+ T cells initially play a critical role in the expansion of the immune response. After encountering an APC that expresses the specific antigenic peptide–MHC II pairing, the CD4+ T cell can then signal back to the APC to elicit factors that allow for CD8+ T cell activation. This process is accomplished via expression of specific costimulatory molecules and the release of certain cytokines. This so-called licensing of CD8+ T cells for cytotoxic function is a key step within the immune response. This partly describes how CD4+ T cells become helper cells. More recently, there has been further elucidation of their cellular differentiation into well-defined specific Th subsets. Two distinct Th populations have been described, based on their pattern of cytokine synthesis—Th1 cells induce a cell-mediated response whereas Th2 cells promote a humoral response (Fig. 26-14). These two distinct populations differ in their pattern of cytokine synthesis.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


