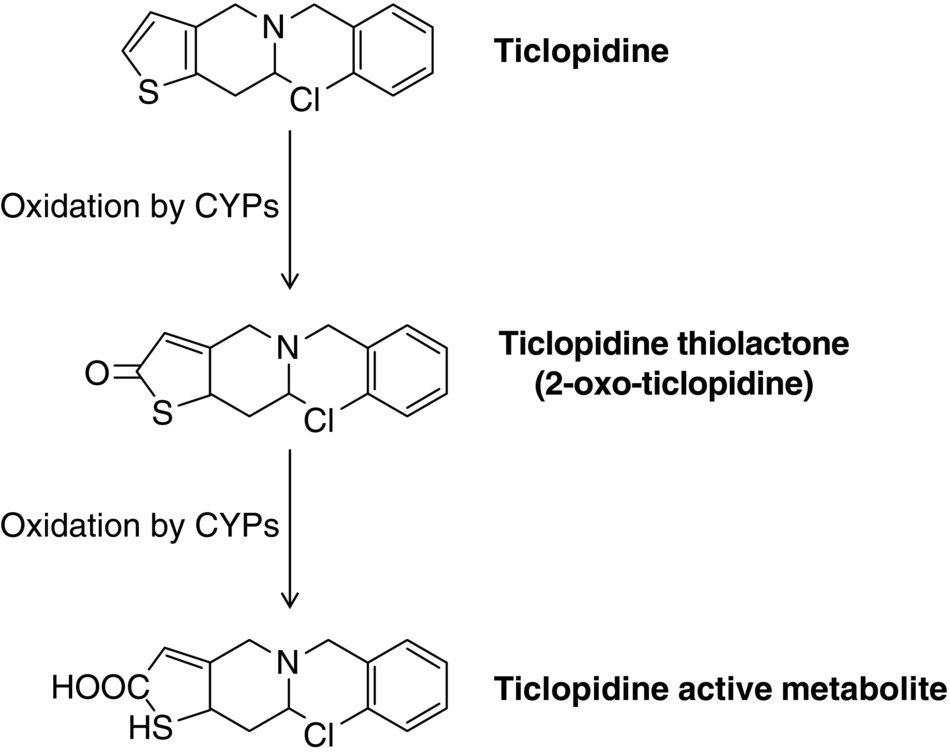
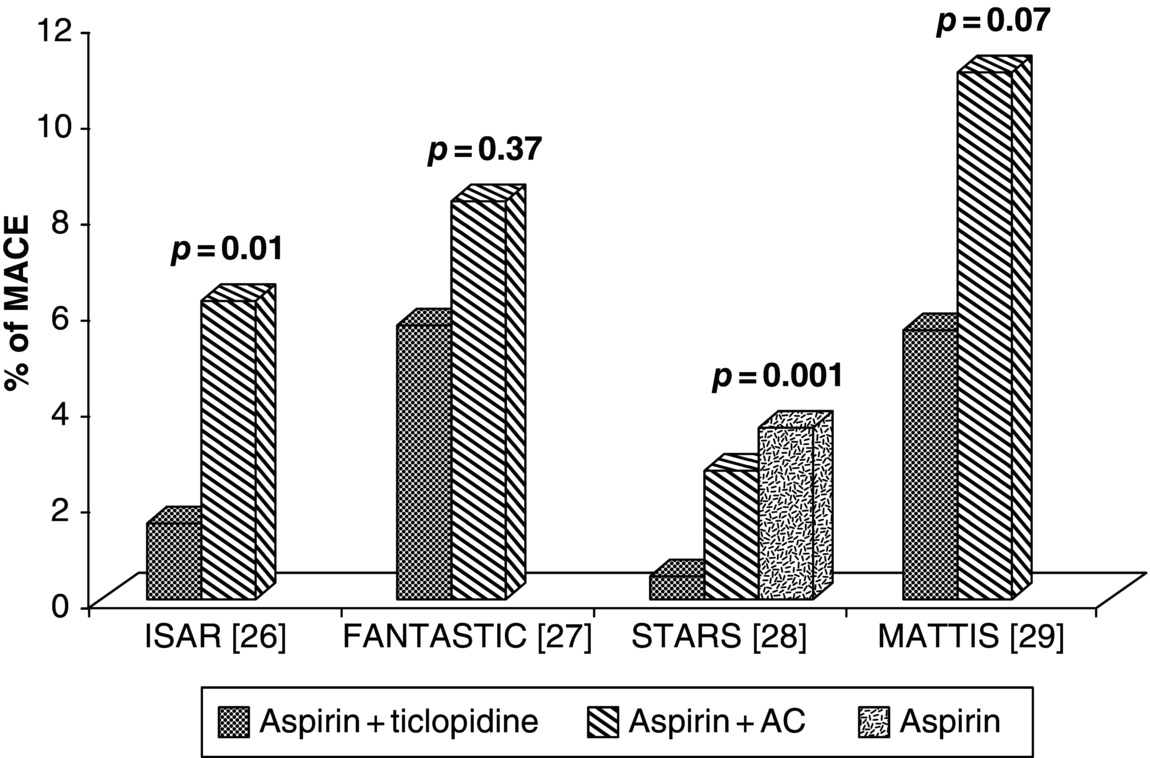
18 Fabiana Rollini, Francesco Franchi, Ana Muñiz-Lozano, and Dominick J. Angiolillo University of Florida College of Medicine, Jacksonville, FL, USA Dual antiplatelet therapy (DAPT) with aspirin and a P2Y12 receptor inhibitor is the cornerstone of treatment to decrease ischemic events in patients with acute coronary syndrome (ACS) or undergoing percutaneous coronary intervention (PCI) [1]. Thienopyridines (ticlopidine, clopidogrel, and prasugrel) selectively and irreversibly inhibit the P2Y12 receptor. However, these are prodrugs that require hepatic conversion into their active metabolite [2]. The first clinically available thienopyridine was ticlopidine, which was approved by the Food and Drug Administration for clinical use in the early 1990s and as adjunctive therapy to aspirin after stent implantation in 2001 [3]. Several studies have demonstrated the efficacy of ticlopidine as monotherapy in patients with cerebrovascular disease or with unstable angina [4, 5, 6]. The Canadian American Ticlopidine Study (CATS) trial showed a 30% reduction in the incidence of recurrent stroke, vascular death, and myocardial infarction (MI) in patients with recent thromboembolic stroke randomized to ticlopidine 500 mg/day (vs. placebo) [4]. The Ticlopidine Aspirin Stroke Study (TASS) trial compared ticlopidine with aspirin in patients with a history of transient ischemic attack, amaurosis fugax, or minor stroke showing a significant benefit with ticlopidine therapy [5]. Moreover, ticlopidine was previously considered the drug of choice for DAPT with aspirin in patients after coronary artery stenting to prevent stent thrombosis [7]. However, nowadays, the role of ticlopidine has become marginal due to its numerous and potentially severe side effects and the development of safer and more effective antiplatelet agents [8, 9]. This chapter will provide an overview on pharmacological properties and clinical development of ticlopidine. Ticlopidine [5-(2-chlorophenyl)methyl-4,5,6,7-tetrahydrothieno[3,2-c] pyridine], a first-generation thienopyridine, is an oral antagonist of the adenosine diphosphate (ADP) receptor, which acts by irreversible blockade of the P2Y12 platelet surface receptor [10]. Ticlopidine does not inhibit ADP-induced platelet aggregation in vitro, because it is a prodrug that requires hepatic biotransformation into an active metabolite [11]. Thirteen metabolites have been described and cytochrome P450 (CYP)2C19 and CYP2B6 are known to be involved in ticlopidine metabolism. However, the complete metabolic pathway producing the active thiolactone, which binds irreversibly the P2Y12 receptor by generation of 2-oxo-ticolpidine, is still unclear (Figure 18.1) [12]. Figure 18.1 Ticlopidine metabolism. CYP, cytochrome P450. The in vivo onset of action after oral administration of ticlopidine (250 mg bid) occurs after 24–48 h, while about 5 days are required to reach maximal ADP-induced platelet inhibition. Approximately 85% of the drug is absorbed and the peak plasma concentration is reached after 2 h from drug intake. Bioavailability is increased by food and decreased by antacids. After repeated dosing, the clearance of ticlopidine decreases significantly, probably due to the inhibition on its own metabolism by inhibiting CYP2C19 and CYP2B6. Elimination of ticlopidine is 60% renal and 23% gastrointestinal. Platelet inhibitory effects are saturated at a dosage of 250 mg twice a day; a single 500 mg daily dose was also tested and showed a faster full inhibitory effect (4 days) but at the expense of more gastrointestinal side effects [7, 13, 14]. To date, few studies have been published on the pharmacodynamic (PD) effects of ticlopidine compared with other thienopyridines. Although platelet inhibition is dose dependent, the relation between the degree of platelet inhibition and plasma concentrations of active metabolites is still unclear [15]. Maximum platelet inhibition occurs 3–5 days after daily administration in healthy subjects, and the offset occurs 3–4 days after discontinuation of 250 mg daily doses and 11–13 days after 500 mg doses [14]. The addition of ticlopidine to aspirin allows reducing platelet reactivity more than what each single drug does [16], even in patients undergoing PCI (ADP (2 μM)-induced platelet aggregation: 36 ± 12 in aspirin plus ticlopidine versus 54 ± 12 in aspirin alone; p < 0.0001) [17]. Same results were found also using vasodilator-stimulated phosphoprotein (VASP), which is an assay specific to evaluate the P2Y12 receptor signaling [18]. PD effects of ticlopidine were also evaluated in patients with chronic kidney disease, but no significant difference compared with healthy volunteers was shown; thus, no dose adjustment is required in patients with impaired renal function [19]. In a study on patients with stable angina scheduled for coronary angiography, the administration of a ticlopidine 1500 mg loading dose showed significantly higher platelet inhibition at 24 h compared to the modest antiplatelet effect achieved by the standard dosage (250 mg twice daily), as assessed by ADP-induced platelet fibrinogen binding. However, at 1 week, no difference was found between the two strategies [20]. It is well known that CYP2C19 polymorphisms reduce the antiplatelet effect of clopidogrel and increase the risk of ischemic cardiac events, such as stent thrombosis [21].Ticlopidine is a prodrug metabolized by multiple CYPs, including CYP2C19 as well [11]. However, in vivo production of ticlopidine active metabolites and pharmacokinetic properties do not depend mainly on CYP2C19 activity. Moreover, CYP2C19 polymorphisms did not show to reduce ticlopidine antiplatelet therapy [22]. Maeda et al. demonstrated that ADP (20 µmol/L)-induced platelet aggregation, evaluated by light transmittance aggregometry, was similar in extensive, intermediate, and poor metabolizers (PM), according to CYP2C19 genotypic status. Furthermore, platelet aggregation was significantly lower in PM patients on ticlopidine treatment group than on clopidogrel (33.8% vs. 19.1%; P < 0.001), and in clopidogrel PM that switched to ticlopidine, platelet aggregation was further suppressed (33.4% vs. 17.3%; P < 0.01) [23]. The role of ticlopidine in patients with pharmacological resistance to clopidogrel was tested in a case report series [24]. However, this was more adequately evaluated by Campo et al. who studied 143 patients undergoing PCI for stable coronary artery disease or ST-segment elevation MI [25]. The authors found that patients with resistance to clopidogrel (21%) were mainly responsive to ticlopidine and vice versa. In particular, 83% of patients who were clopidogrel nonresponders resulted to be responsive to ticlopidine, reaching a significant higher level of platelet inhibition (ADP (20 µmol/L)-induced platelet aggregation: 69 ± 15 vs. 44 ± 18; P < 0.05). Only 3.5% of patients (5 of 143) demonstrated both clopidogrel and ticlopidine resistance, suggesting a drug-specific mechanism, rather than a class effect, for thienopyridine resistance. Although some studies have assessed the role of ticlopidine in the other settings of vascular disease manifestations [4, 5], ticlopidine has been mostly acknowledged for its impact in the setting of patients undergoing PCI and stenting (Figure 18.2). In fact, despite the introduction of coronary stents in the late 1980s, the optimal antithrombotic treatment regimen to prevent stent thrombosis remained a challenge, hampering the clinical application of stent procedures. The combination of aspirin with anticoagulants was associated with high rates of thrombotic and bleeding complications. Therefore, an approach to achieve synergistic antiplatelet effects by blocking two key pathways, cyclooxygenase-1 with aspirin and the ADP receptor with ticlopidine, introducing the concept of “DAPT,” was approached in four pivotal trials. Figure 18.2 Rate of major adverse cardiac events (MACE) in the studies comparing ticlopidine plus aspirin to anticoagulant therapy plus aspirin. AC, anticoagulant therapy. The Intracoronary Stenting and Antithrombotic Regimen (ISAR) trial evaluated the effect of ticlopidine after PCI and coronary stenting [26]. Patients (n = 517) were randomized to receive DAPT with aspirin (100 mg twice a day) plus ticlopidine (250 mg twice a day started immediately after the procedure and continued for 4 weeks) or conventional therapy with intravenous heparin, vitamin K antagonist (VKA), and aspirin. The primary cardiac end point was a composite of cardiovascular death, MI, aortocoronary bypass surgery, or repeated PCI of the stented vessel. At a 30-day follow-up, ticlopidine significantly reduced the incidence of the primary end point (1.6% vs. 6.2%; RR, 0.25; 95% CI, 0.06–0.77; p = 0.01) with an 82% reduction of MI. The rate of hemorrhagic events was also reduced in the ticlopidine group (p < 0.001). In the Full Anticoagulation Versus Aspirin and Ticlopidine (FANTASTIC) study, patients (n = 485) undergoing stent implantation were randomized to receive DAPT with ticlopidine (500 mg loading dose administered immediately after procedure and then 250 mg bid for 6 weeks) plus aspirin (100–325 mg for life) or anticoagulant therapy (heparin and VKA) for 6 weeks plus aspirin [27]. The primary end point, the occurrence of any bleeding or peripheral vascular complications during the 6 weeks after stent implantation, was significantly reduced in the ticlopidine arm (13.5% vs. 21%; OR, 0.59; 95% CI, 0.39–0.98; p
Ticlopidine
Pharmacological properties
Pharmacodynamic profile
Genetic considerations
Clinical efficacy in patients undergoing PCI
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


